


Beyond Blood Sugar: Rethinking Insulin Resistance
- S A

- May 19, 2024
- 14 min read
Updated: Aug 5, 2025
Part 1
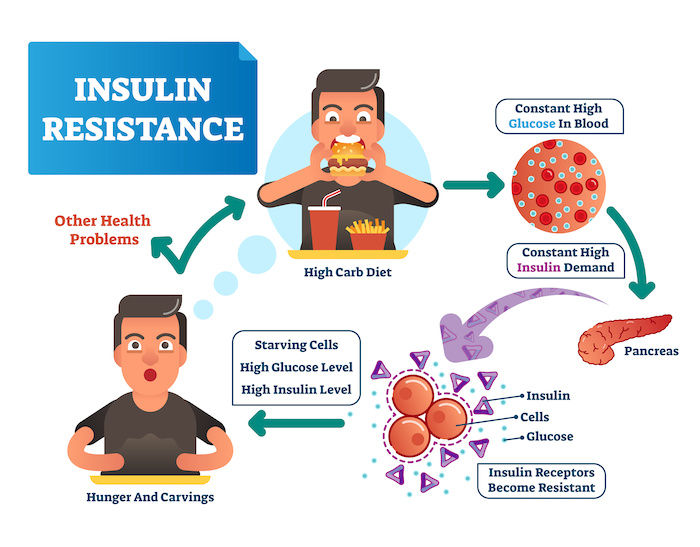
For many, achieving good health often boils down to one number: blood sugar. But what if there's a bigger picture to consider? This blog dives into the concepts of insulin resistance and metabolic health, exploring the crucial role they play in our overall well-being.
Today, we delve deeper into the complexities of insulin resistance, specifically addressing a crucial paradigm shift that's essential for effectively managing this condition.
The Flawed Glucose-Centric View: A Missed Opportunity
Traditionally, the medical field has primarily focused on blood sugar levels when diagnosing and managing insulin resistance. While maintaining healthy blood sugar is undoubtedly important, it's vital to recognize the bigger picture.

Credit: Freepic - Vectorjuice
The Canary in the Coal Mine: Elevated Insulin as an Early Warning
At the core of the issue lies a simple fact: insulin resistance, in its early stages, is often characterized by elevated insulin levels in the blood, not necessarily elevated blood sugar. The body, in a commendable effort to compensate for insulin resistance, ramps up insulin production to keep glucose levels in check. Imagine a car engine working harder to maintain speed with a faulty transmission – that's what's happening here.
Here's where the traditional approach falls short:
Elevated insulin levels can rise significantly (up to 20 years!) before glucose levels become abnormal. This elevated insulin serves as an early warning sign of impending trouble, a canary in the coal mine that often goes unnoticed due to our hyper-focus on glucose.
By solely focusing on glucose, we potentially miss the chance to intervene early and prevent the progression of insulin resistance and its downstream consequences. Early detection and intervention are crucial for managing any health condition, and insulin resistance is no exception.
Insulin Resistance: When Cellular Communication Breaks Down
Imagine insulin as a messenger, knocking on the door of your cells, like a muscle/fat cell, to deliver glucose, the fuel your cells need for energy. In a healthy scenario, the doors readily open, and the cells efficiently absorb the glucose upon insulin's instruction.
However, over time, due to various factors we'll explore later, these cellular doors become less responsive. The insulin "knocks" might be heard faintly, or maybe the door opens just a crack. This is the essence of insulin resistance – cells become resistant to insulin's message.

Credit: Micoope
Important Nuances: Selective Resistance and Elevated Insulin
It's crucial to note that insulin resistance isn't a uniform phenomenon. Not all cells are affected equally. Some cells lose their sensitivity to insulin's message, while others continue to respond normally. This is very important to keep in mind, as this phenomenon leads to a host of metabolic conditions.
Here's another key point often overlooked: insulin resistance is almost always accompanied by elevated blood insulin levels, a condition called hyperinsulinemia. Think of it as two sides of the same coin.
Hyperinsulinemia: A Compensatory Response (with Unintended Consequences)
When some cells become resistant, the pancreas, the organ that produces insulin, ramps up production to compensate. This leads to higher levels of insulin circulating in the bloodstream – hyperinsulinemia.
Here's where things get interesting. While some cells are resistant, others remain insulin-sensitive. These overstimulated cells experience an exaggerated response to the elevated insulin levels, essentially getting more than they need.
Glucose Transporter | Location | Insulin Dependence | Primary Function | GlucoseAffinity | Impact of Glucose Concentration |
GLUT1 | Throughout the body (red blood cells, brain, kidneys) | Insulin-independent | Basal glucose uptake, ensures steady supply | High | Significant impact on transport rate. GLUT1 is nearly saturated at normal blood glucose concentrations, but increased glucose levels can further enhance uptake. |
GLUT2 | Liver, intestines, kidneys | Insulin-independent | Liver function, intestinal sugar absorption, kidney glucose reabsorption | Low | Significant impact on transport rate. At lower glucose concentrations, GLUT2 plays a role in reabsorption (kidneys) or uptake (liver). At higher concentrations, transport increases. |
GLUT3 | Brain, placenta (focally), Ovaries | Partially Insulin-dependent | Glucose uptake for brain energy, possible role in fetal development (placenta) | Very High | Less impact on transport rate due to extremely high affinity. However, extremely low glucose levels can still limit uptake. |
GLUT4 | Skeletal muscle, adipose tissue (fat cells) | Insulin-dependent | Major transporter for muscle and fat cell glucose uptake, energy storage | Med | Significant impact on transport rate. In the absence of insulin, GLUT4 is inactive regardless of glucose concentration. With insulin, glucose concentration influences uptake considerably. |
Impact on Hyperinsulinemia
In conditions like insulin resistance (e.g., type 2 diabetes), some cells, particularly muscle and fat cells, become less responsive to insulin's signal. This means GLUT4 translocation is impaired, and glucose uptake by these tissues decreases.
To compensate for the decreased glucose uptake, the pancreas increases insulin production, leading to hyperinsulinemia (high blood insulin levels).
While some cells are resistant, others like those expressing GLUT1 (e.g., red blood cells) can still be responsive to high insulin levels. This can lead to excessive glucose uptake in these tissues, even though they might not necessarily need it.
What Does 'High Affinity for Glucose' Mean in the Context of GLUT Transporters?
"Affinity" refers to how easily a transporter binds and moves glucose at low concentrations.
High affinity = can transport glucose even when blood glucose is low (e.g. brain).
Low affinity = needs higher blood glucose levels before it starts working efficiently (e.g. liver, pancreas).
The Outcome: A Dysfunctional System
The net result of insulin resistance and hyperinsulinemia is a dysfunctional system. Cells that need glucose aren't getting it efficiently, while others are overreacting to excess insulin.
GLUT4-expressing tissues (muscle, adipose) become insulin resistant→ Can’t take up glucose efficiently→ Leads to high blood sugar and hyperinsulinaemia
GLUT1/GLUT3-expressing tissues (brain, RBCs, cancer cells) are insulin-independent→ Still get glucose freely, even in insulin resistance
GLUT2 (liver, pancreas) also becomes dysfunctional in insulin resistance, especially in the pancreas, where glucose sensing and insulin release are impaired over time
Why This Creates Metabolic Chaos (Organ-Specific Downstream Effects)
Brain
Glucose uptake in the brain is insulin-independent, via GLUT1 (astrocytes, endothelial cells) and GLUT3 (neurons).
These are high-affinity transporters — so the brain always gets glucose, even during fasting or hypoglycaemia.
But: the brain does respond to insulin in non-metabolic roles, especially in the:
Hypothalamus (appetite regulation, energy sensing)
Hippocampus (memory and cognition)
Olfactory bulb (smell-based feeding cues)
So insulin in the brain is not for glucose entry, but for signalling and communication.
What Does Insulin Normally Do in the Brain?
In a healthy, insulin-sensitive brain:
Insulin in the hypothalamus suppresses appetite and increases energy expenditure.
It enhances leptin sensitivity, promoting satiety.
It modulates dopamine reward pathways to reduce food-seeking behaviour.
It regulates glucose sensing and nutrient partitioning.
Also, insulin helps with:
Memory and synaptic plasticity in the hippocampus.
Protecting neurons from oxidative stress and inflammation.
What Happens in Brain Insulin Resistance?
Even though glucose still enters the brain, insulin signalling is blunted in key brain regions.
This has major effects:
a. Appetite Dysregulation
Insulin fails to suppress appetite → hyperphagia, especially for carbs and fat.
Leptin resistance develops → satiety signals ignored.
Increased ghrelin and NPY/AgRP signalling → hunger ramped up.
Reward circuits (dopamine) become hypersensitive to food cues → food addiction patterns.
b. Energy Imbalance
The brain no longer senses that the body has enough energy stored.
Drives a feed-forward loop: eat more, move less, store more fat.
c. Cognitive Effects
Insulin resistance in the hippocampus impairs memory and learning.
Associated with Alzheimer’s disease — sometimes called “Type 3 diabetes”.
Promotes neuroinflammation, oxidative stress, and mitochondrial dysfunction.
d. Neurovascular Impact
Insulin resistance contributes to small vessel disease, poor blood flow, and endothelial dysfunction.
Increases risk for stroke, vascular dementia, and cognitive decline.
Systemic Feedback Loops Between Brain and Body
Adipose insulin resistance → leptin resistance → brain can’t sense energy reserves
Liver insulin resistance → high glucose → brain exposed to hyperglycaemia
Brain insulin resistance → hyperinsulinaemia (pancreas overcompensates)
Hyperinsulinaemia → worsens peripheral insulin resistance
Brain drives more food intake → worsens the cycle
Analogy: The Broken Thermostat
Think of the brain as the thermostat of the body’s energy system:
Normally, it detects when the "room" (body) has enough energy and shuts off the "furnace" (appetite, insulin production).
But in insulin resistance, the thermostat is broken.
It thinks the room is cold even when it's hot.
So it cranks up the furnace — more eating, more fat storage, more insulin.
Meanwhile, the other rooms (liver, fat, muscle) are overheating and malfunctioning.
Downstream Effects of Brain Insulin Resistance
Effect | Systemic Outcome |
Appetite dysregulation | Overeating, especially ultra-processed foods |
Leptin resistance | Loss of satiety, increased fat storage |
Increased insulin output | Hyperinsulinaemia, β-cell stress |
Reward sensitivity | Cravings, food addiction |
Neurodegeneration | Memory loss, Alzheimer's, dementia risk |
Reduced energy expenditure | Sedentary behaviour, further fat gain |
Adipose Tissue
In healthy adipose tissue, insulin is a storage hormone:
Insulin promotes:
Glucose uptake via GLUT4 (→ used for fat synthesis and glycerol backbone of TGs)
Lipogenesis (creating triglycerides from FFAs and glucose)
LPL (lipoprotein lipase) activity at the capillary wall→ pulls in TGs from chylomicrons/VLDL for storage
Insulin suppresses:
Lipolysis: breaks down stored triglycerides into FFAs and glycerol→ by inhibiting hormone-sensitive lipase (HSL)
In short:
Fed state + insulin = fat storage, not fat release
What Happens in Adipose Insulin Resistance?
Now the story flips.
Insulin loses its ability to suppress lipolysis
Fat cells become leaky — they dump FFAs into the bloodstream even when insulin is high
Insulin-driven glucose uptake is impaired
Less GLUT4 activity → less glucose enters fat cells→ Reduced glycerol-3-phosphate → less TG synthesis→ More FFAs escape
Impaired LPL signalling
Less fat storage from incoming VLDL/chylomicrons
Lipids stay in circulation longer → remnant cholesterol, TGs, atherogenic lipoproteins
Systemic Consequences of Adipose Insulin Resistance
a. FFA Spillover
Excess FFAs go to liver, muscle, pancreas→ promote lipotoxicity, ectopic fat storage, organ insulin resistance
b. Hepatic Overload
Liver takes up the FFAs → makes more VLDL → fatty liver + dyslipidaemia
c. Pancreatic β-cell stress
FFAs impair insulin secretion → β-cell dysfunction, type 2 diabetes
d. Muscle insulin resistance
FFAs interfere with insulin signalling → muscle can't take up glucose
e. Inflammation
Adipocytes release inflammatory cytokines (TNF-α, IL-6, resistin)→ Recruit macrophages → vicious inflammatory loop → worsens insulin resistance
f. Adipokine Imbalance
↓ Adiponectin (insulin-sensitising)
↑ Leptin resistance (brain can't sense fullness properly)
Mechanisms That Drive Adipose Dysfunction
a. Overfilled Fat Cells
Once fat cells reach storage limit → become hypoxic, inflamed, dysfunctional→ “Adipose tissue expandability” theory: when fat can’t expand, fat spills elsewhere (viscera, liver, muscle)
b. Visceral vs Subcutaneous Fat
Visceral fat (around organs) is more metabolically active and pro-inflammatory→ Stronger driver of insulin resistance than subcutaneous fat
c. Hyperinsulinaemia
Chronic high insulin tries to suppress lipolysis, but eventually fails→ Constant fat storage + spillover → fat redistribution and metabolic stress
Key Markers of Adipose Dysfunction
Marker | What It Indicates |
High fasting insulin | Compensatory insulin to suppress leaky fat cells |
Elevated FFAs | Uncontrolled lipolysis |
Low adiponectin | Loss of insulin sensitivity |
High leptin + leptin resistance | Brain ignores satiety signals |
TG/HDL ratio > 3.5 (mg/dL) | Proxy for insulin resistance |
Waist circumference | Visceral adiposity (metabolically dangerous fat) |
Analogy
Imagine fat tissue as a warehouse:
Normally, insulin is the manager who directs fat to be stored safely inside.
In insulin resistance, the workers stop listening:
They start leaking fat out the back door (FFAs)
They don’t accept new shipments (VLDL)
They send inflammatory messages to other organs
Meanwhile, the liver, muscles, and pancreas are forced to store fat they’re not designed to hold → breaking the system.
Liver
The liver uses GLUT2, which is:
Insulin-independent
Bidirectional (glucose can flow in and out)
Low affinity (only active at high glucose levels — i.e. after meals)
So, glucose entry into liver cells does not require insulin.
But insulin still plays a critical role in what the liver does with that glucose once inside.
What Does Insulin Normally Do in the Liver?
Insulin binds to insulin receptors on hepatocytes and:
Inhibits:
Gluconeogenesis (new glucose production)
Glycogenolysis (glycogen breakdown)
Ketogenesis
Stimulates:
Glycogen synthesis (glucose storage)
De novo lipogenesis (fat creation from excess carbs)
VLDL export (packaging excess fat into lipoproteins)
So, even though glucose can enter without insulin, insulin directs glucose’s fate inside the liver.
What Happens in Hepatic Insulin Resistance?
Insulin resistance in the liver means:
Insulin's ability to suppress glucose production is impaired→ The liver keeps making and releasing glucose into the blood→ Even when blood glucose is already high
But insulin’s stimulation of fat production (DNL) and VLDL export may remain intact or even exaggerated→ So you get fatty liver, hypertriglyceridaemia, and remnant cholesterol buildup
This is sometimes called "selective insulin resistance":
The liver ignores insulin's instruction to stop making glucose,but still listens to its command to turn sugar into fat.
Downstream Effects of Hepatic Insulin Resistance
a. Hyperglycaemia
Liver pumps out glucose 24/7
Adds to elevated fasting blood sugar
Key driver of prediabetes and type 2 diabetes
b. Hyperinsulinaemia
Pancreas keeps pumping out insulin trying to suppress hepatic glucose output
c. Hepatic Steatosis (Fatty Liver)
Excess FFAs from adipose and DNL in liver get packaged into triglycerides
Stored in liver (steatosis) or exported as VLDL → high TG, small dense LDL
d. Atherogenic Dyslipidaemia
High triglycerides
Low HDL
Remnant cholesterol accumulation → promotes atherosclerosis
e. Impaired Ketogenesis
Insulin suppresses ketone production — even when glucose is not being cleared
So the person may have energy shortage in tissues (e.g. brain) despite high glucose
The Clinical Hallmarks of Hepatic Insulin Resistance
Marker | What it Means |
High fasting glucose | Liver is still making glucose despite insulin |
High fasting insulin | Pancreas is trying to compensate |
High triglycerides | Excess fat from liver (VLDL) |
Fatty liver on imaging (NAFLD) | DNL and FFA overload |
TG/HDL ratio > 3.5 (mg/dL units) | Marker of insulin resistance |
ApoB elevated | More atherogenic lipoproteins (VLDL remnants, LDL) |
Analogy
Think of the liver as a factory with two departments:
Glucose Department: supposed to shut down when insulin shows up.
Fat Department: supposed to only ramp up when energy is in excess.
In insulin resistance, the Glucose Department ignores orders and keeps running,but the Fat Department overreacts and works overtime.
Result: Too much glucose in the blood, too much fat in the liver and blood, and a confused metabolic system.
Ovaries — Insulin and Androgen Production (PCOS)
Insulin and Aromatase Activity
Aromatase is the enzyme that converts testosterone into oestrogen.
Insulin stimulates aromatase activity — especially in adipose (fat) tissue.
Therefore, high insulin (often in the context of insulin resistance or obesity) can:
Increase conversion of androgens to oestrogens (especially oestrone, a weaker form)
Lead to oestrogen dominance in both men and women
In men, this can contribute to gynecomastia, low libido, and reduced testosterone
In women, especially postmenopausal, this can worsen oestrogen-related symptoms
Insulin and Testosterone in Women (e.g. PCOS)
In women, insulin has the opposite effect in the ovaries:
High insulin directly stimulates thecal cells in the ovaries to produce more androgens (e.g. testosterone)
Simultaneously, insulin suppresses Sex Hormone Binding Globulin (SHBG) from the liver→ This leads to more free (active) testosterone
This is a central mechanism in PCOS:
High insulin → High androgens → Anovulation, acne, hirsutism, irregular periods
Insulin and Ovulation
Normal insulin signalling supports ovulation via:
Sensitising the brain (hypothalamus) to regulate GnRH, LH, and FSH appropriately
Supporting follicular development and maturation
Chronic hyperinsulinaemia impairs this finely tuned process:
Disrupts LH/FSH balance, leading to irregular ovulation or anovulation
Excess androgens from ovarian thecal cells block proper follicle development
Leads to the polycystic appearance of ovaries (many immature follicles stuck in limbo)
Key concept
Selective insulin sensitivity: Ovaries still respond to insulin (make androgens), even when muscle/adipose do not (can’t take in glucose). This selective effect worsens PCOS.
Intestines — GLP-1, Nutrient Sensing, and Lipoprotein Handling
GLUT Transporters
Enterocytes use SGLT1 (sodium-glucose transporter) and GLUT2.
GLUT2 is insulin-independent, but insulin resistance can upregulate SGLT1 and alter glucose absorption dynamics.
Incretins (GLP-1, GIP)
Intestines sense glucose and secrete GLP-1 and GIP, which enhance insulin release.
Chronically high insulin/glucose → GLP-1 resistance in β-cells and hypothalamus.
Lipid Transport
Intestinal cells package dietary fat into chylomicrons (similar to VLDL but ApoB48 instead of ApoB100).
In insulin resistance, chylomicron clearance is delayed, contributing to postprandial hyperlipidaemia.
Consequences
Excess nutrient absorption, impaired satiety signalling.
Sluggish chylomicron clearance → atherogenic remnant cholesterol buildup.
GLP-1 signalling fatigue → poorer insulin control, increased appetite.
Androgens - Insulin’s Role in Their Overproduction Beyond the Ovary
Adrenal Glands
Insulin can enhance adrenal androgen production (e.g. DHEA) under stress.
Chronic hyperinsulinaemia interacts with ACTH (adrenocorticotropic hormone) to dysregulate the HPA axis.
Liver
Low SHBG in insulin resistance → more free (bioactive) testosterone and estrogen.
Liver also fails to deactivate androgens/estrogens properly in NAFLD.
Increased IGF-1 Production: Hyperinsulinemia can also lead to increased IGF-1 production by the liver. IGF-1 can further stimulate androgen production in the ovaries, contributing to the hormonal imbalance in PCOS.
Hyperinsulinemia and elevated IGF-1 are considered key contributors to metabolic syndrome. They can promote insulin resistance, fat storage, and inflammation, all of which play a role in the development of this condition.
Muscle
Can’t take up glucose efficiently→ Less glycogen storage→ Energy shortfall→ Glucose lingers in blood
Pancreas
Initially overproduces insulin in response to glucose→ Eventually β-cell burnout and Type 2 diabetes onset
Insulin Resistance: Multi-System Effects Across the Body
Tissue/Organ/System | Normal Insulin Action | Insulin Resistance Effects | Downstream Systemic Consequences |
Liver | - Suppresses gluconeogenesis - Stimulates glycogen & fat synthesis - VLDL packaging | - Gluconeogenesis persists - Fat production continues - VLDL overproduction | - Hyperglycaemia - Fatty liver (NAFLD) - Atherogenic dyslipidaemia |
Muscle | - Glucose uptake via GLUT4 - Glycogen synthesis | - Impaired glucose uptake - Reduced glycogen storage | - Elevated postprandial glucose - More insulin demand - Fatigue |
Adipose Tissue | - Glucose uptake - Suppresses lipolysis - Promotes TG storage | - FFA spillover - Inflammation - Reduced expandability | - Lipotoxicity - Visceral fat accumulation - Elevated TGs |
Brain | - Appetite regulation - Enhances leptin & insulin signalling - Cognitive support | - Appetite not suppressed - Reward pathways dysregulated - Cognitive decline | - Overeating - Obesity - Alzheimer’s risk - Depression |
Ovaries | - Modest support for steroidogenesis | - Increased androgen output - Still sensitive to insulin | - PCOS - Infertility - Hirsutism - Elevated testosterone |
Pancreas | - Senses glucose (GLUT2) - Releases insulin | - Initially overcompensates - Later fails | - Hyperinsulinaemia → β-cell burnout → T2D |
Intestines | - GLP-1/GIP secretion - Nutrient sensing - Fat absorption via chylomicrons | - GLP-1 resistance - Increased SGLT1 - Slow chylomicron clearance | - Poor satiety - Post-meal hyperlipidaemia |
Kidneys | - Inhibits gluconeogenesis - Regulates sodium & glucose reabsorption | - ↑ SGLT2 → ↑ glucose retention - Sodium retention | - Fasting hyperglycaemia - Hypertension - Diabetic nephropathy |
Immune System | - Promotes anti-inflammatory profile (M2 macrophages) - Maintains immune regulation | - Chronic inflammation (M1 macrophages) - Cytokine storm (IL-6, TNF-α) | - Worsened insulin resistance - Atherosclerosis - Autoimmune activation |
Cardiovascular System (Endothelium) | - Increases NO (nitric oxide) for vasodilation - Protects endothelium - Inhibits inflammation | - ↓ NO production - ↑ oxidative stress & endothelial dysfunction | - Hypertension, arterial stiffness - Plaque formation (atherosclerosis) - Increased CVD risk |
Hormonal Systems (Cortisol, Thyroid, GH, Sex Hormones) | - Regulates interactions with insulin to coordinate energy usage - Cortisol helps in stress - T3 increases glucose oxidation | - ↑ Cortisol (stress, poor sleep) → worsens IR - ↓ T3 (low thyroid) → lowers energy burning - GH may antagonise insulin | - Worsened insulin resistance - Fat gain (esp. visceral) - Low energy, cold intolerance - Disrupted menstrual cycles or libido |
Skin | - Supports skin repair - Normal hair growth and collagen balance | - Poor wound healing - Acanthosis nigricans (velvety, dark skin folds) - Skin tags | - Early visible marker of insulin resistance - Higher infection risk - Impaired tissue regeneration |
The Vicious Loop
Let’s tie it all together:
Insulin resistance → hyperinsulinaemia
→ muscle/adipose resist glucose uptake
→ liver keeps producing glucose and fat
→ ovaries overproduce androgens
→ intestines overabsorb nutrients
→ GLP-1 and leptin signalling blunted
→ more hunger, more storage, more dysfunction
Analogy
Think of the body as a company where insulin is the CEO:
Some departments (muscle, adipose) stop listening to orders.
But other departments (ovaries, intestines) follow orders too well and start overproducing things (androgens, chylomicrons).
The result is organisational chaos — systems that were supposed to be coordinated now amplify metabolic disorder.
The Paradigm Shift: A Focus on Insulin as a Marker
A shift in perspective is needed. By acknowledging the limitations of the glucose-centric view and recognizing the crucial role of insulin, we can move towards a more comprehensive understanding of insulin resistance. Here's why insulin deserves more attention:
Insulin levels can provide a valuable window into cellular communication and fuel usage within the body. By monitoring insulin levels, we gain a deeper understanding of how effectively insulin is working and how well your body is utilizing glucose for energy.
Focusing on insulin as a marker allows for earlier detection of insulin resistance, paving the way for preventative measures and lifestyle changes. Early intervention is key to preventing the progression of insulin resistance and its associated health risks.
Moving Beyond: Inadvertently Worsening the Problem
In some cases, our current approach to managing insulin resistance can have unintended consequences. Here's how:
Some treatment options, when blood sugar finally becomes uncontrolled, involve increasing insulin levels through injections or medications (sulphonylureas). While this might lower glucose in the short term, it can exacerbate the underlying insulin resistance in the long run.
This focus on lowering blood sugar through increased insulin creates a vicious cycle. The more insulin is pushed, the worse the insulin resistance becomes. This cycle can contribute to the development of various chronic diseases.
The Takeaway: A Broader Perspective is Key
By acknowledging the limitations of the glucose-centric view and recognizing the crucial role of insulin, we can move towards a more comprehensive understanding of insulin resistance. Focusing on insulin as a marker and implementing lifestyle changes that address the root causes become essential for effectively managing and preventing this condition.
Looking Ahead: Solutions on the Horizon
In future discussions, we'll delve deeper into the factors contributing to insulin resistance, explore effective lifestyle strategies, and shed light on the connection between insulin resistance and various chronic diseases. Remember, knowledge is power. By equipping yourself with a deeper understanding of insulin resistance, you can take control of your metabolic health and pave the way for a healthier future.


Comments