


Pathophysiology of Atherosclerosis: How Heart Disease Really Begins - Part 2
- S A

- Aug 15, 2025
- 12 min read
Updated: Feb 3
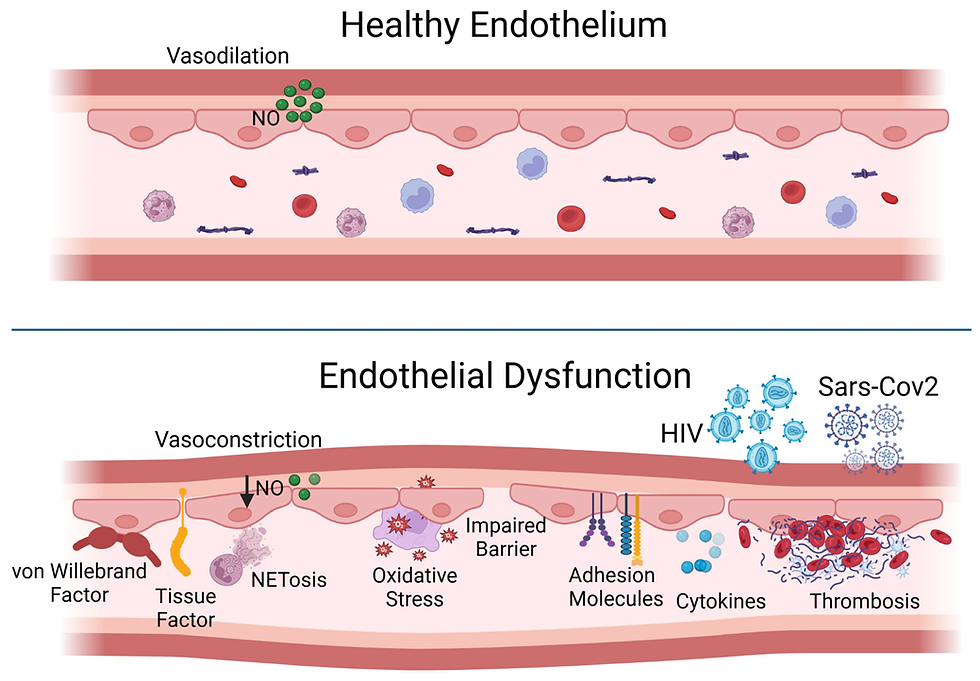
In the first part of this series, we saw how atherosclerosis starts with a breach in the endothelial barrier — the “first crack in the wall” — followed by the infiltration of LDL particles into the arterial lining. Once there, these lipoprotein visitors are no longer in the fast-flowing, antioxidant-rich environment of the bloodstream. Instead, they’re lodged in a relatively stagnant, enzyme-rich space where trouble can quietly brew.
This is where the real transformation begins. Isolated from normal defences, LDL undergoes chemical changes that alter its structure and behaviour. Polyunsaturated fats in its shell become vulnerable to oxidative attack, its protein backbone can be modified, and it takes on a form the immune system no longer recognises as “self.”
In this next step, we’ll explore how LDL becomes oxidised and chemically trapped within the arterial wall — and why this shift is the trigger that turns a passive deposit into an active instigator of inflammation and plaque growth.
Step 3: How LDL Becomes Oxidised and Trapped in the Arterial Wall
Now that we learned the difference between lbLDL and sdLDL and why the latter is more prone to get trapped, let’s take a deeper dive into how LDL becomes oxidised, why this matters, and what happens next in the arterial wall:
LDL Enters the Subendothelial Space
Normally, LDL circulates in the blood delivering cholesterol and lipids to cells.
Under endothelial dysfunction (caused by high BP, inflammation, smoking, etc.), the endothelial barrier becomes more permeable.
LDL particles — especially small, dense ones — slip beneath the endothelium into the subendothelial space (intima layer of the artery).
Retention of LDL in the Arterial Wall
Once inside, LDL particles bind to proteoglycans in the extracellular matrix, especially if they are small and dense.
This retention is key — trapped LDL is now exposed longer to damaging influences.
Think of it as a piece of food stuck between your teeth — it’s more likely to rot there than if it were swept away.
Oxidative Modification (Oxidation) of LDL
In the subendothelial space, LDL is not protected by antioxidants like it is in plasma.
It gets exposed to reactive oxygen species (ROS) from:
Activated immune cells (macrophages, neutrophils)
Endothelial and smooth muscle cells under stress
Two key enzymes play a central role in initiating and accelerating LDL oxidation:
Myeloperoxidase (MPO)
What releases it? Activated neutrophils and macrophages in sites of inflammation — like an artery wall under stress.
How it works:
MPO takes hydrogen peroxide (H₂O₂) (itself a byproduct of oxidative bursts in immune cells) and uses chloride ions to make hypochlorous acid (HOCl).
HOCl is like industrial bleach at a molecular scale — it doesn’t just “oxidise” gently; it aggressively chlorinates and modifies molecules.
Targets in LDL:
Surface phospholipids: PUFA tails on the LDL surface phospholipids are oxidised, forming lipid hydroperoxides.
ApoB-100 protein: Chlorination of tyrosine and oxidation of methionine/histidine residues distorts the protein’s structure, meaning LDL receptors can’t recognise it properly. This pushes LDL towards scavenger-receptor uptake by macrophages.
Lipoxygenase (LOX)
What uses it? Primarily macrophages, endothelial cells, and smooth muscle cells in vascular tissue.
How it works:
LOX is an iron-containing enzyme that directly adds oxygen to cis,cis-1,4-pentadiene structures in PUFA — especially linoleic acid (C18:2, ω-6).
This generates lipid hydroperoxides (LOOH) — unstable, reactive molecules.
These hydroperoxides decompose into reactive aldehydes like 4-hydroxynonenal (4-HNE) and malondialdehyde (MDA).
Aldehydes can cross-link proteins, forming a kind of molecular “superglue” that locks ApoB-100 into a damaged, non-functional state.
The Chain Reaction Effect
Once either MPO or LOX damages the surface PUFA or ApoB-100, the oxidation cascade spreads:
Surface damage → core damage: Oxidised phospholipids generate free radicals that burrow into the LDL core, oxidising the cholesteryl esters.
Structural damage: ApoB-100 modifications prevent LDL recycling by the liver’s LDL receptor pathway, increasing LDL’s residence time in plasma — giving it more opportunities to penetrate arterial walls.
Why the body does this in the first place
MPO and LOX evolved as antimicrobial weapons — perfect for attacking bacterial cell walls, which also contain PUFA-like structures.
The problem: LDL’s lipid shell looks chemically similar to a bacterial membrane. So in the context of chronic inflammation (from insulin resistance, smoking, hypertension), immune cells keep firing at LDL — a case of friendly fire.
Analogy:Picture LDL as a cargo truck:
The paint and outer shell = surface phospholipids with PUFA tails.
The driver’s ID badge = ApoB-100 (needed for docking at liver “warehouses”).
MPO is like someone splashing industrial bleach on the truck — it eats away at the paint, corrodes metal, and melts the driver’s ID badge so he’s no longer allowed in the main depot.
LOX is like a precise but destructive cutting torch — it slices straight into the vulnerable metal seams, producing sparks that ignite cargo inside.
Once the outer damage starts, the cargo (PUFA-rich cholesteryl esters) becomes rancid, and the truck is diverted to a “junkyard” (macrophages in the arterial wall), where it adds to the growing pile of wreckage (atherosclerotic plaque).

Image Credit: Researchgate
Why sdLDL ends up with more PUFA while large buoyant LDL (lbLDL) has more MUFA/SFA
1. The “aging” and remodelling effect
When LDL circulates longer (as sdLDL tends to), it is continuously remodelled by enzymes like lecithin–cholesterol acyltransferase (LCAT), cholesteryl ester transfer protein (CETP), and hepatic lipase.
CETP swaps cholesteryl esters in LDL for triglycerides from VLDL/HDL. Many of those cholesteryl esters in VLDL and HDL are PUFA-rich (because dietary PUFA tends to get packaged into them).
Over time, sdLDL accumulates more linoleic acid and other PUFA in its cholesteryl ester core.
2. Dietary fat composition shows up differently in LDL sizes
Large LDL particles are usually cleared faster and are more reflective of the liver’s initial lipid output, which often has higher MUFA/SFA content (especially if the diet is lower in seed oils and higher in SFA/MUFA).
sdLDL forms downstream from TG-rich VLDL — and those VLDL are enriched with PUFA if the person eats a PUFA-heavy diet or is insulin resistant.
This is because PUFA, especially omega-6, preferentially go into triglycerides and cholesteryl esters in the liver.
The "Preferential Loading"
Our liver is an incredibly efficient sorter of fuel. However, it has a specific biochemical "affinity" for Linoleic Acid (LA).
The Enzyme Magnet: The enzymes responsible for building triglycerides (like DGAT) and cholesteryl esters (like ACAT2) actually have a higher binding affinity for Polyunsaturated Fatty Acids (PUFAs) compared to Saturated Fats (SFAs).
The Disposal Logic: Saturated fats are the body’s preferred "clean-burning" fuel. The body often tries to burn SFAs for immediate energy or store them in stable adipose tissue. Because PUFAs are chemically unstable and potentially toxic in high amounts, the liver tries to "get rid of them" by packing them into VLDL ships and sending them out into the blood as fast as possible.
The Insulin Resistance Multiplier
If you add Insulin Resistance to a PUFA diet, the problem grows exponentially:
The Flood: In insulin resistance, your fat cells "leak" fatty acids back to the liver constantly.
The Overproduction: The liver, overwhelmed by this flood of leaked fat and high insulin, pumps out massive amounts of VLDL.
The Chain Reaction: More VLDL means more "Handshakes." More handshakes mean more "stolen" stable cargo and more "collapsed" sdLDL arsonists.
The liver is a master packer, but it has a fatal flaw: it prefers to ship out PUFAs as quickly as possible. When you eat seed oils, your liver packs your VLDL 'trucks' with high-octane Omega-6. Through the 'Handshake of Death,' this volatile cargo is transferred to your LDL, causing them to collapse into small, dense 'arsonists.' You aren't just dealing with small particles; you are dealing with particles that have been biologically 'rigged' to fail the moment they leave the liver
3. Why PUFA enrichment matters for oxidation
PUFA have multiple double bonds → very susceptible to lipid peroxidation.
The higher PUFA content in sdLDL’s phospholipid coat and CE core makes it chemically easier for reactive oxygen species to start the oxidation chain reaction.
Large buoyant LDL, with more MUFA/SFA, is more stable because those fatty acids are harder to oxidise.
In short:
Formation path: TG-rich VLDL → CETP swapping → hepatic lipase trimming
Result: Higher PUFA content in sdLDL core & coat
Consequence: Greater oxidation risk → stronger link to atherogenesis
Seed oils rich in linoleic acid (LA), such as soybean, corn, sunflower, safflower, and canola oil, provide the exact substrate (PUFAs) that enzymes like lipoxygenase (LOX) and myeloperoxidase (MPO) use to oxidise LDL particles.
Let’s unpack why this matters, both biochemically and practically
Linoleic Acid (LA) Is Highly Oxidation-Prone
LA is a polyunsaturated omega-6 fatty acid, and because of its multiple double bonds, it is chemically unstable and highly susceptible to lipid peroxidation.
When LDL contains high levels of LA in its lipid layer (as it often does in people consuming seed oil-rich diets), it becomes an easy target for oxidation via:
LOX directly oxidising LA within LDL
MPO generating reactive species that further attack these unstable lipids
This leads to lipid hydroperoxides and aldehydes, which not only damage LDL but also create inflammatory byproducts like 4-HNE (4-hydroxynonenal) — a potent oxidative stress marker that can disrupt cellular function and DNA.
Seed Oils Alter LDL Composition
When diets are high in seed oils, the fatty acid composition of LDL particles shifts. The LDL lipid membrane becomes enriched in linoleic acid, making it:
More likely to oxidise
More likely to be retained in the arterial wall
More likely to stimulate immune responses
In contrast, saturated fat–rich LDL tends to contain more monounsaturated and saturated fatty acids, which are much more resistant to oxidation.
Oxidised LA Metabolites (OXLAMs) Are Harmful
When LA is oxidised (either during cooking, processing, or in the body), it produces oxidised linoleic acid metabolites (OXLAMs) — such as:
9-HODE and 13-HODE
4-HNE
These have been shown to:
Increase endothelial dysfunction
Promote macrophage foam cell formation
Impair mitochondrial function
Trigger chronic inflammation and insulin resistance
Human & Animal Studies Support This Mechanism
The Minnesota Coronary Experiment (1970s) — showed that replacing saturated fat with seed oils lowered cholesterol but increased mortality, particularly from cardiovascular causes.
Sydney Diet Heart Study — a reanalysis showed that higher linoleic acid intake increased the risk of death from heart disease despite lowering LDL.
Modern observational studies suggest that while omega-6 fats in whole-food forms (e.g., nuts/seeds) may not pose issues, refined seed oils — especially when heated — are linked to increased markers of inflammation and oxidation.
Bottom Line
Seed oils rich in linoleic acid feed the very oxidative pathways that convert benign LDL into dangerous oxLDL — particularly in the context of:
Insulin resistance
Low antioxidant status
Chronic inflammation
Highly processed, calorie-dense diets
If LDL is the passenger, then linoleic acid is the explosive cargo, and enzymes like MPO and LOX are the spark.
From Metabolic Syndrome to Molecular Sabotage
1. Metabolic Syndrome Sets the Stage: Metabolic syndrome brings together insulin resistance, hyperglycaemia, hypertriglyceridaemia, hypertension, and chronic low-grade inflammation.
Insulin resistance keeps VLDL production high → more triglyceride-rich LDL → more small, dense LDL (sdLDL).
Hyperglycaemia and high free fatty acids promote endothelial stress and immune cell activation.
2. Immune System Gets Trigger-Happy
Endothelial cells in insulin resistance express more adhesion molecules (VCAM-1, ICAM-1), calling in monocytes.
Monocytes → macrophages in the arterial intima → release inflammatory cytokines (IL-1β, TNF-α, IL-6).
These cytokines upregulate MPO and LOX expression in vascular tissue.
3. Oxidation Risk Skyrockets
MPO upregulation: More HOCl production, more ApoB-100 chlorination, faster phospholipid damage.
LOX upregulation: Higher PUFA oxidation rates, more lipid hydroperoxide and reactive aldehyde production.
4. The “Perfect Storm” for oxLDL Formation. You now have:
More LDL particles in circulation (ApoB ↑).
More of those particles being small and dense (easier arterial entry).
More oxidative enzymes ready to attack PUFA tails and ApoB-100.
Reduced antioxidant defence (low vitamin E, glutathione depletion, low paraoxonase activity on HDL).
The result: accelerated conversion of native LDL → oxidised LDL, which gets stuck in arterial walls and drives foam cell formation.
Analogy
Imagine a city with:
More trucks on the road (↑ ApoB).
Smaller trucks that fit into alleyways (sdLDL).
Cargo that spoils easily (PUFA-rich cholesteryl esters).
More vandals with blowtorches and bleach (↑ LOX, ↑ MPO).The odds of a “truck wreck” in the wrong place go way up.
PUFAs - The Evolutionary Baseline vs. The Modern Load
When we look at the sheer volume of PUFA intake, the argument shifts from "is this molecule toxic?" to "can the biological system handle this load?" For the vast majority of human evolution, linoleic acid (LA) intake was roughly 1% to 2% of total calories, coming from whole foods like walnuts, animal fats (from pasture-raised animals), and seeds.
Today: In Canada and the US, that number has skyrocketed to 15% to 20%+ of total calories.
The Biological "Storage" Problem: Unlike carbohydrates (stored as glycogen) or proteins (used for structure), PUFAs are integrated directly into your cell membranes and adipose tissue.
The Half-Life: The half-life of linoleic acid in human fat cells is approximately 2 years. This means if you stop eating seed oils today, your "volatile cargo" remains in your "tankers" for years to come.
The "Clean Diet" Paradox
Even if you follow a "clean, non-UPF" diet, if you are using "healthy" oils like Olive oil or even large amounts of Poultry (which is high in LA due to soy/corn feed), you are still hitting a PUFA threshold that human physiology has never encountered.
Cardiolipin Oxidation: Cardiolipin is a phospholipid in the inner mitochondrial membrane that is essential for energy production. It is highly enriched with linoleic acid. When LA intake is excessive, cardiolipin becomes highly susceptible to oxidation, which can lead to mitochondrial "leaks" and decreased metabolic efficiency.
Prostaglandin Overdrive: While the body regulates the conversion of LA to Arachidonic Acid (AA), having a massive reservoir of LA in the cell membranes means the "pre-loaded" substrate for inflammation is always at maximum capacity.
The "Quality Fat" Hierarchy
This brings us to why people who dive deep into the physiology and biochem, which is what I tend to focus on, eventually move toward a hierarchy of fats. If the goal is to prevent the "oxidative fire" in the arterial wall, the strategy becomes:
Saturated Fats (SFA): The "Shield." Zero double bonds. Chemically inert and impossible to peroxidize. (Think Tallow, Ghee, Coconut Oil).
Monounsaturated Fats (MUFA): The "Buffer." One double bond. Very stable and generally neutral-to-protective. (Think Extra Virgin Olive Oil, Macadamia).
Polyunsaturated Fats (PUFA): The "Spice." Multiple double bonds. Necessary in small amounts (Omega-3/6 balance), but dangerous as a primary fuel source.
The human body is an expert at managing oxidative stress in small doses. We have endogenous antioxidants like Glutathione and Superoxide Dismutase to put out small "sparks."
The problem with the 15-20% intake is that it creates a perpetual state of oxidative demand. You eventually deplete your antioxidant reserves. Once those reserves are gone, the MPO and LOX enzymes have free rein to attack the LDL particles, regardless of how "clean" the rest of your diet is.
Is it "Purely Physiology" that Seed Oils are Bad?
If we define "bad" as "increasing the statistical probability of molecular damage beyond the body's natural capacity to repair it," then yes—the massive escalation of PUFA intake is physiologically reckless.
Even in a "perfect" person, you are increasing the "volatility" of your biological tissues. The "Quantity" of the load eventually breaks the "Quality" of the system.
Conclusion — The Turning Point: From Lipid Delivery to Inflammatory Crisis
Once LDL slips beneath the endothelium, the stage is set for a catastrophic transformation. Cut off from the antioxidant-rich protections of the bloodstream, the particle's internal cargo—now heavily enriched with polyunsaturated fatty acids (PUFAs) due to a century-long, 75x increase in seed oil consumption—becomes a volatile liability. In this stagnant environment, Myeloperoxidase (MPO) and Lipoxygenase (LOX) act as molecular saboteurs, striking the "weak hinges" of the linoleic acid tails to ignite a chain reaction of lipid peroxidation.
This isn't just a matter of "bad luck"; it is a matter of dosage and duration. The massive evolutionary mismatch of our modern PUFA intake ensures that our LDL "tankers" are loaded with highly reactive fuel that the body's natural "cleanup crews" (like Vitamin E and Glutathione) were never designed to manage at such scale. This process produces Oxidized Linoleic Acid Metabolites (OXLAMs)—toxic breakdown products that act as chemical beacons, signaling the immune system to attack what it once recognized as a vital delivery vehicle.
In the context of modern metabolic dysfunction—marked by high triglycerides and insulin resistance—this crisis is amplified. Elevated triglycerides ensure that these particles are remodeled into small, dense LDL (sdLDL), which linger in the blood longer and penetrate the arterial wall with ease. What began as a routine mission to deliver cholesterol for cellular repair has been subverted by a combination of high-pressure "traffic," metabolic "potholes," and "explosive cargo."
We have moved from a simple lipid transport mission to a state of chronic, immunological alarm embedded deep within the arterial wall. In our final part, we will follow this chain reaction to its grim conclusion: how the immune system’s desperate attempt to sequester these "rancid" particles leads to the formation of necrotic plaque, the structural failure of the arterial wall, and the sudden, life-threatening clotting response that characterizes the modern heart attack.
📢 A Note on "Living Science"
Science is not a static destination; it is a moving target. While the principles of Turnover, Signaling, and Tension are grounded in decades of metabolic research, new peer-reviewed data emerges every day.
I am committed to accuracy. If you are a researcher, clinician, or dedicated student of physiology and you find a piece of data here that does not align with the latest high-quality evidence, please reach out. I welcome civil, evidence-based corrections. My goal is to keep this resource as the most accurate "No-Nonsense" guide to Cardio Vascular Health on the internet. Let’s get better together.
*Disclaimer:
The information provided in this blog is for educational and informational purposes only and should not be construed as medical advice. While every effort is made to ensure accuracy, the content is not intended to replace professional medical consultation, diagnosis, or treatment. Always seek the guidance of a qualified healthcare provider with any questions regarding your health, medical conditions, or treatment options.
The author is not responsible for any health consequences that may result from following the information provided. Any lifestyle, dietary, or medical decisions should be made in consultation with a licensed medical professional.
If you have a medical emergency, please contact a healthcare provider or call emergency services immediately.

Comments