


Pathophysiology of Atherosclerosis: How Heart Disease Really Begins - Part 1
- S A

- Aug 15, 2025
- 12 min read
Updated: Feb 3
In our previous blogs, we challenged the long-standing narrative that saturated fat and high LDL cholesterol are the primary culprits behind heart disease. We explored how LDL isn’t inherently harmful, how not all LDL is created equal, and how metabolic context — not cholesterol alone — determines cardiovascular risk.
We also highlighted that small, dense LDL particles (not just LDL-C) are more atherogenic, and that their formation is often a consequence of insulin resistance, excess carbohydrate intake, and chronic inflammation — not saturated fat in a vacuum.
Now, it's time to zoom inside the arteries and look at what’s actually happening on a cellular and biochemical level. Because heart disease doesn’t begin with cholesterol — it begins with a damaged endothelium, a dysregulated immune response, and a metabolic environment that allows otherwise useful lipoproteins to become toxic.
This blog unpacks the true pathophysiology of atherosclerosis — step-by-step — to help you understand:
Why LDL shows up at the scene of the crime
What transforms helpful lipoproteins into harmful agents
How inflammation, oxidative stress, and metabolic dysfunction initiate plaque formation
And most importantly, how we can interrupt this process with smarter dietary and lifestyle interventions
Let’s take a deep dive into how heart disease really begins — and why it’s not just about your cholesterol numbers.
What Is Atherosclerosis?
Atherosclerosis — often simplified as “clogged arteries” — is far more than just a build-up of cholesterol. It’s a dynamic, decades-long process rooted in inflammation, endothelial dysfunction, oxidative stress, and lipoprotein trafficking gone awry.
It’s characterised by the accumulation of lipids, immune cells, and fibrous tissue, which together form plaques that narrow and stiffen arteries. Over time, these plaques can rupture, leading to heart attacks or strokes.
But contrary to conventional wisdom, it’s not just about high cholesterol. Let’s start where the problem actually begins.
Step 1: Endothelial Dysfunction — The First Crack in the Wall
The endothelium is a thin, protective layer lining the inside of your blood vessels. It regulates blood flow, nutrient exchange, and keeps the vessel wall smooth and anti-inflammatory.
However, metabolic stress and other insults can damage this lining. Key culprits include:
High blood pressure: Constant elevated pressure physically stretches and stresses the arterial walls, leading to microscopic injuries in the endothelium. This makes it more permeable to LDL particles and inflammatory cells. Over time, it reduces the elasticity and integrity of the vessels, priming them for plaque development.
Smoking/Pollution: Cigarette smoke introduces reactive oxygen species (ROS) and other toxins into the bloodstream, directly causing oxidative damage to endothelial cells. It also reduces nitric oxide (NO) availability, impairing vasodilation and promoting inflammation.
Chronic inflammation: Persistent low-grade inflammation (often driven by poor diet, stress, obesity, or infection) causes the immune system to be on high alert. Cytokines like TNF-alpha and IL-6 can make the endothelium more adhesive, attracting monocytes and LDL into the vessel wall — the first step of plaque formation.
High blood sugar/insulin resistance: Elevated glucose promotes the formation of advanced glycation end-products (AGEs), which stiffen the blood vessels and impair endothelial function. Insulin resistance also reduces nitric oxide production and increases oxidative stress — both of which further damage the endothelium.
Oxidised LDL particles: LDL itself isn’t the enemy, but when it becomes oxidised (due to ROS, poor antioxidant status, or a damaged metabolic state), it becomes highly inflammatory. OxLDL triggers immune activation and is readily taken up by macrophages — a key event in plaque development.
Low nitric oxide production: Nitric oxide is critical for keeping blood vessels relaxed, flexible, and anti-inflammatory. Factors like poor sleep, low physical activity, low sun exposure, or high oxidative stress reduce NO production, leading to vasoconstriction, platelet aggregation, and endothelial inflammation.
Chronically High Insulin Levels
Hyperinsulinaemia, often seen in insulin resistance and metabolic syndrome, contribute to inflammation, reduced nitric oxide (NO) production, and endothelial dysfunction:
Insulin and Inflammation
Chronically high insulin stimulates pro-inflammatory pathways, especially the NF-κB signalling pathway.
It increases the production of cytokines like IL-6, TNF-α, and CRP, which promote systemic low-grade inflammation.
This inflammatory environment makes the endothelium more reactive and encourages monocyte adhesion to the vessel wall — an early step in plaque formation.
Insulin and Nitric Oxide (NO)
Under normal conditions, insulin actually promotes NO production via the PI3K-Akt-eNOS pathway, which helps vasodilation and endothelial health.
But in insulin resistance, this pathway becomes impaired, while a competing one — the MAPK pathway — becomes dominant. This shift:
Reduces NO production
Increases endothelin-1, a potent vasoconstrictor
The result is a pro-constrictive, pro-inflammatory vascular environment.
Insulin and Endothelial Dysfunction
Reduced NO means impaired vasodilation, which is a hallmark of endothelial dysfunction.
High insulin also promotes oxidative stress by increasing ROS production.
It stimulates vascular smooth muscle proliferation, increases clotting factors, and promotes LDL retention in the vessel wall.
In summary: High insulin shifts the vascular environment from relaxed and anti-inflammatory to tense, inflamed, and sticky — greatly increasing the risk of atherosclerosis over time.
Once this protective layer is disrupted, it becomes “sticky” — allowing immune cells and lipoproteins to penetrate the arterial wall. Together, these factors make the normally smooth and protective endothelial surface leaky, sticky, and inflamed — setting the stage for LDL particles to enter, become oxidised, and kick off the cascade of atherosclerosis.
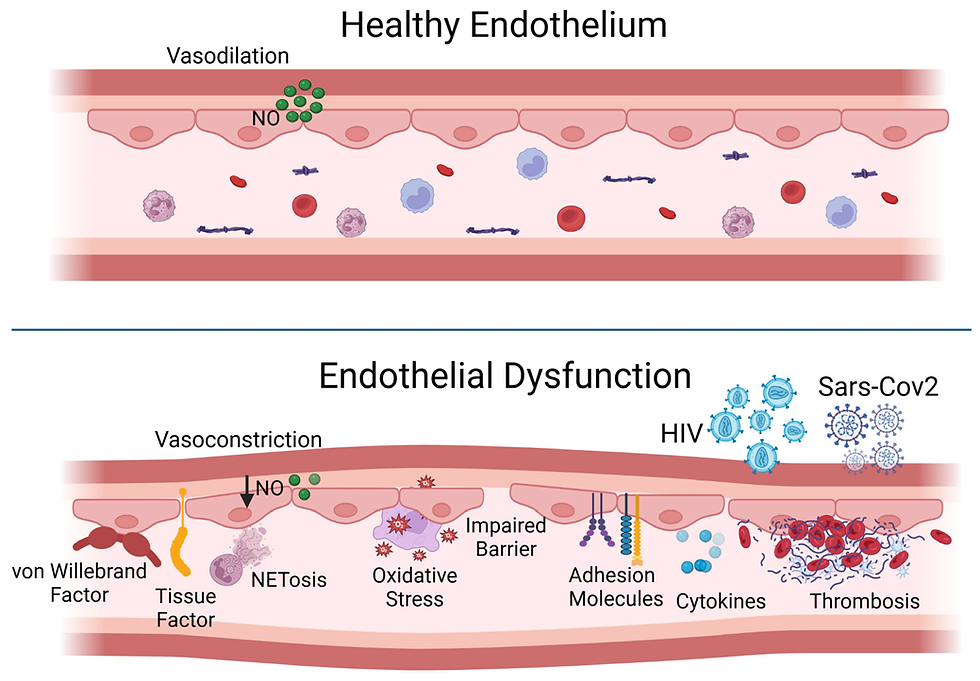
Image Credit: MDPI
Step 2: LDL Enters the Arterial Wall
When the endothelium is damaged, LDL particles (especially small, dense LDL) slip beneath the surface and lodge in the intima (the innermost arterial layer).
Importantly, it’s not just any LDL that’s the problem — it’s excess sdLDL particles, especially in the context of:
Oxidative stress
Inflammation
Poor metabolic health
Impaired LDL clearance (e.g., low thyroid, insulin resistance)
How VLDL Becomes Small, Dense LDL
VLDL particles are released from the liver to deliver triglycerides to tissues. As they circulate, lipoprotein lipase (LPL) removes triglycerides, shrinking VLDL into IDL (intermediate-density lipoprotein) and eventually into LDL. In a metabolically healthy state, many of these LDL particles are large and buoyant. But in insulin resistance or high-triglyceride states, cholesterol-poor LDL particles undergo further triglyceride enrichment (via CETP) and subsequent hepatic lipase action, producing small, dense LDL (sdLDL). These sdLDL particles are more likely to penetrate the arterial wall, bind to proteoglycans in the intima, and undergo oxidation — making them particularly atherogenic.
let’s map the whole cascade from high triglycerides to oxidised LDL and plaque formation, so the chain makes sense in one continuous metabolic story.
Step 1: High Triglycerides – the trigger
High-carb, high-sugar diet (↑ hepatic DNL → ↑ VLDL output)
Insulin resistance (↓ LPL activity → slower TG clearance)
Excess calorie intake (regardless of source, but carbs drive it harder)
Liver pumps out large, TG-rich VLDL particles.
Step 2: LCAT action in HDL
Lecithin–cholesterol acyltransferase (LCAT) is an enzyme in the bloodstream, mostly active on HDL.
It takes free cholesterol (FC) from cell membranes or chylomicron remnants, and esterifies it — attaching a fatty acid from phosphatidylcholine (lecithin) → creating a CE (Cholesteryl ester).
This CE is tucked away in HDL’s core.
Step 3: CETP transfer
Cholesteryl ester transfer protein (CETP) swaps those CEs from HDL into apoB-containing lipoproteins (VLDL, IDL, LDL) in exchange for triglycerides.
This is how LDL ends up with most of its CE cargo.
The type of fatty acid in the CE reflects the dietary fat pattern — more PUFA in diet → more PUFA in CEs; more saturated/MUFA → more stable CEs.
Step 4: VLDL shrinking → LDL formation
VLDL from the liver carries triglycerides and CEs. As lipoprotein lipase (LPL) removes triglycerides for tissues, VLDL becomes IDL, then LDL.
The CE proportion increases as triglycerides are depleted, making LDL mostly a CE carrier.
Why sdLDL is a problem
Compared to large, buoyant LDL:
Smaller → can slip through the arterial endothelium more easily.
Denser → less buoyant, stays in circulation longer (↑ residence time).
TG-depleted surface often means altered phospholipid makeup → more prone to oxidative modification.
Oxidation vulnerability
sdLDL is more likely to get oxidised because:
Longer circulation time → more exposure to oxidative stress.
Lower antioxidant content (vitamin E, carotenoids) compared to large LDL.
Modified surface lipids make apoB more exposed and easier to attack.
From oxidised LDL to atherogenesis
Oxidised LDL (oxLDL) is recognised by macrophage scavenger receptors, not LDLR.
Macrophages engulf oxLDL → foam cells.
Foam cells pile up in the intima → fatty streaks → fibrous plaques.
Chronic inflammation, smooth muscle migration, and extracellular matrix changes → atherosclerosis.
Analogy
Imagine LDL as a tanker truck:
Outer shell (phospholipids + free cholesterol) = the truck’s metal body — partly water-friendly (paints/coatings) so it can travel in “watery roads” (blood).
ApoB protein = the driver’s ID badge — without it, the truck can’t dock at delivery sites (LDL receptors).
Core cholesteryl esters = the oil barrels in the truck’s cargo hold — dense, hydrophobic, meant for delivery to sites needing cholesterol.
Triglycerides = perishable goods also inside, but usually offloaded earlier (muscle/fat tissue) before the LDL makes its final cholesterol delivery.
Now — the type of oil in those barrels matters:
PUFA oil = delicate, goes rancid easily → attracts “rust” (oxidation) → truck becomes damaged and more likely to cause accidents (atherosclerosis).
Saturated/MUFA oil = more stable, can handle a long trip without spoiling.
The Chemistry of the "Trade"
Cholesterol, in its "free" form, is a jagged molecule that likes to sit on the surface of membranes. To pack it into the center of a transport ship (HDL or LDL), it must be converted into a Cholesteryl Ester (CE). To make a CE, you have to "esterify" it, which essentially means clamping a fatty acid tail onto the cholesterol molecule.
The Problem: LCAT doesn't just create fatty acids out of thin air. It has to steal one from somewhere.
The Solution: It steals it from Phosphatidylcholine.
The "Scissors and Glue" Action
Phosphatidylcholine (PC) is the most abundant phospholipid in the human body. It is the primary building block of every single cell membrane we own. From our brain cells to your gut lining, we are essentially held together by PC.
Think of a lipoprotein (LDL/HDL) like an Oil Tanker, and Phosphatidylcholine is a phospholipid that makes up the "skin" of your lipoproteins. It has a head and two fatty acid tails.
The Inside: Is hydrophobic (hates water). This is where the fat and Cholesteryl Esters hide.
The Outside: Is in constant contact with your blood, which is mostly water.
You need a "wrapper" that can touch both fat and water. This is the Phosphatidylcholine. It has a "head" that loves water and two "tails" that love fat.
The tails point inward to hold the cargo.
The heads point outward to let the particle float smoothly in the blood.
LCAT acts like a pair of chemical scissors:
The Cut: LCAT snips one of the fatty acid tails off the Phosphatidylcholine molecule.
The Paste: It immediately glues that tail onto a molecule of Free Cholesterol.
The Result: You now have a Cholesteryl Ester (which dives into the center of the ship) and a leftover piece called Lysophosphatidylcholine (which gets recycled).
The Structural Integrity of the Shell (Pillar 3)
Phosphatidylcholine isn't just a donor; it is the primary building block of the LDL Shell.
If the Phosphatidylcholine itself is made of oxidized seed oils, the "skin" of the LDL truck is weak, leaky, and "pre-lit."
If the Phosphatidylcholine is made of stable SFAs and MUFAs, the shell is "Armored."
Think of Phosphatidylcholine as the parts warehouse for your cholesterol transport system. To turn raw cholesterol into stable cargo, the LCAT enzyme has to go to the warehouse and grab a 'fatty acid tail.' If your warehouse is full of cheap, volatile seed oil parts, your LCAT has no choice but to build 'exploding cargo.' If you want stable LDL, you must provide the warehouse with high-quality, stable building blocks like those found in egg yolks and healthy animal fats.
Why this matters for the "Seed Oil" Debate
This is the "Smoking Gun". LCAT’s preferred "donor" tail from Phosphatidylcholine is usually found at the sn-2 position of the molecule.
The Ancestral State: In a healthy, ancestral diet, that sn-2 position is often occupied by a stable fat.
The Modern Sabotage: If you consume high amounts of seed oils, your Phosphatidylcholine becomes "loaded" with Linoleic Acid (PUFA).
The Consequence: When LCAT goes to do its job, it is forced to grab that "kinked," unstable Linoleic Acid tail and glue it to the cholesterol.
Since PC makes up the entire surface of the LDL particle, the quality of the fatty acid tails determines how tough that skin is.
The SFA Skin: If you eat stable fats, the tails are straight and packed tight. This creates a "Leather Jacket" for the LDL—it’s tough, waterproof, and hard to damage.
The PUFA Skin: If you eat seed oils, the tails are "kinked" and loose. This creates a "Tissue Paper" skin. Not only is it easy to tear, but those kinks are the exact spots where Oxygen attacks, starting the fire of oxidation right on the surface of the particle.
This is exactly how you end up with an LDL core full of volatile Oxidized Linoleic Acid instead of stable Saturated Esters. The Phosphatidylcholine is the "Magazine" that feeds the "Gun" (LCAT).
You can’t move cargo without a container. Phosphatidylcholine is the container, and Choline is the raw material. If you aren't eating enough Choline (found richly in egg yolks and liver), your liver effectively 'goes on strike.' The fat stays in the liver (NAFLD), and the few LDL particles you do manage to send out are built with flimsy, cut-rate materials. For those on plant-based diets, this 'Choline Gap' is a silent driver of metabolic dysfunction that no amount of 'low-fat' eating can fix.
Actionable Tip:
Prioritize Choline: If you want stable LDL and a clean liver, you need Phosphatidylcholine. If you don't eat eggs, consider a high-quality Sunflower Lecithin or Alpha-GPC supplement to ensure your liver has the "shipping materials" it needs to keep the Lipid Loop moving.
Why PUFA in the “oil barrel” is the weak link
What’s actually most prone to oxidation in LDL is the fatty acid tail of the cholesteryl ester in the “oil barrel” (and the PUFA in the phospholipid shell). Cholesterol itself — the rigid steroid ring — is remarkably stable. Pure cholesterol won’t just oxidise easily in the body.
Cholesteryl esters = cholesterol + fatty acid.
PUFA fatty acids have multiple double bonds, which are chemically reactive.
Those double bonds are fragile — like “weak hinges” — so free radicals or oxidative enzymes can attack them easily.
When the PUFA tail oxidises, the whole CE molecule changes shape and becomes oxidised LDL (oxLDL).
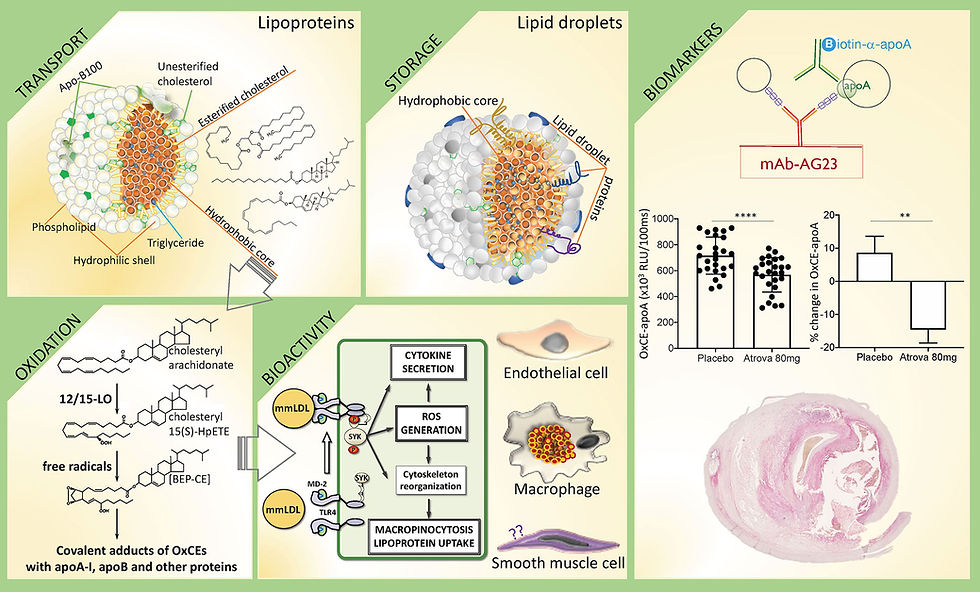
Image Credit: frontiers
So why blame cholesterol?
Because for decades, measurements in arteries and plaques showed “cholesterol-rich” deposits. Here’s how the misunderstanding took hold:
Pathologists found yellow, fatty streaks in arteries and chemical analysis showed high cholesterol content.
Early methods couldn’t easily separate oxidised CE from cholesterol itself.
The narrative became: “cholesterol is clogging arteries” — without distinguishing stable cholesterol from oxidised cholesterol derivatives (oxysterols).
Over time, we’ve learned that it’s modified LDL (oxidised, glycated, PUFA-damaged) that’s inflammatory and dangerous — not native LDL per se.
A better way to phrase the real problem
Atherosclerosis risk is less about how much cholesterol you have in the tankers, and more about:
How fragile their cargo is (PUFA-rich CEs oxidise easily).
How many tankers are on the road at once (ApoB particle count).
How easily they get stranded (small, dense LDL gets trapped in arterial intima).
How quickly cleanup crews (LDL receptors, HDL) can clear them.
How composition changes with diet/metabolism
High PUFA diet → PUFA-rich CEs → more double bonds → more oxidation-prone LDL.
High saturated or MUFA diet → more stable CEs (palmitic, stearic, oleic acids) → LDL less susceptible to oxidative damage.
Insulin resistance / high TG → CETP activity rises, more PUFA-rich TG exchange, smaller LDL, higher oxidation risk.
The full chain
High TG
→ Large TG-rich VLDL
→ CETP swaps CE for TG in LDL/HDL
→ TG-rich LDL
→ Hepatic lipase trims → sdLDL
→ ↑ Arterial penetration + ↑ Oxidation susceptibility
→ OxLDL uptake by macrophages
→ Foam cell formation
→ Plaque growth.
Feature | Large, Buoyant LDL (lbLDL) | Small, Dense LDL (sdLDL) |
Diameter | ~22–25 nm | ~18–20 nm |
Density | ~1.019–1.044 g/mL | ~1.044–1.063 g/mL |
Triglyceride content | Low–moderate (already TG-lean when formed) | Very low (TG removed during shrinkage) |
Cholesteryl ester content | High | High (same or slightly higher % by mass after TG removal) |
Protein (ApoB) | Same per particle, but less dense effect because of larger volume | Same per particle, but takes up more % of total mass → higher density |
Buoyancy | High (lower density, more TG) | Low (denser, less TG) |
Formation pathway | Comes from TG-lean VLDL → LDL without major shrinkage | Comes from TG-rich VLDL → hepatic lipase removes TG → particle shrinks |
Metabolic clearance | Cleared more efficiently by LDL receptors | Longer circulation time (less receptor affinity) |
Oxidation susceptibility | Lower (less time in circulation, thicker phospholipid coat) | Higher (longer circulation, smaller size → can penetrate endothelium more easily, thinner coat) |
Atherogenic potential | Lower | Higher |
Why sdLDL is denser
Triglycerides are bulky and low density (~0.9 g/mL).
Cholesteryl esters and ApoB are denser (~1.05–1.3 g/mL).
When TGs are removed by hepatic lipase, you keep most of the dense material but lose the light stuff → particle density increases.
The particle also physically shrinks, so the same amount of ApoB + cholesterol ester is now crammed into a smaller volume.
Key point: Density comes from the loss of TG plus the unchanged protein & cholesterol ester load, not from adding anything new.
Conclusion — From Crack to Cargo
The story of atherosclerosis begins not with fat floating aimlessly in the blood, but with injury to the endothelium — the delicate, single-cell lining that keeps our arteries smooth, responsive, and resistant to unwanted intruders. When this layer becomes dysfunctional through mechanical stress, toxins, inflammation, or metabolic imbalance, it loses its ability to regulate vessel tone, control permeability, and keep the blood’s contents at bay.
This breach opens the door for LDL particles — especially smaller, denser ones — to slip into the subendothelial space. Once inside, they are effectively trapped, unable to return to circulation. Here, in this hidden niche beneath the endothelium, LDL is now cut off from the antioxidant defences of the plasma, leaving it vulnerable to chemical attack.
In the next section, we’ll see how this trapped cargo changes character — oxidised, remodelled, and ultimately transformed into a dangerous trigger for immune activation. This is the pivotal step that turns a structural problem into a full-blown inflammatory and immune-driven disease process.
📢 A Note on "Living Science"
Science is not a static destination; it is a moving target. While the principles of Turnover, Signaling, and Tension are grounded in decades of metabolic research, new peer-reviewed data emerges every day.
I am committed to accuracy. If you are a researcher, clinician, or dedicated student of physiology and you find a piece of data here that does not align with the latest high-quality evidence, please reach out. I welcome civil, evidence-based corrections. My goal is to keep this resource as the most accurate "No-Nonsense" guide to Cardio Vascular Health on the internet. Let’s get better together.
*Disclaimer:
The information provided in this blog is for educational and informational purposes only and should not be construed as medical advice. While every effort is made to ensure accuracy, the content is not intended to replace professional medical consultation, diagnosis, or treatment. Always seek the guidance of a qualified healthcare provider with any questions regarding your health, medical conditions, or treatment options.
The author is not responsible for any health consequences that may result from following the information provided. Any lifestyle, dietary, or medical decisions should be made in consultation with a licensed medical professional.
If you have a medical emergency, please contact a healthcare provider or call emergency services immediately.


Comments