


LDL Cholesterol: Friend, Foe, or Misunderstood? Rethinking Heart Disease in the Context of Modern Nutrition
- S A

- Aug 8, 2025
- 17 min read
Updated: Jan 22
LDL — three letters that have come to symbolise heart disease risk. For decades, the medical and nutritional consensus has been simple: the higher your LDL cholesterol, the greater your risk of cardiovascular events. This line of thinking has shaped dietary guidelines, fuelled statin prescriptions, and turned saturated fat into a global scapegoat.
But is the story that straightforward? Emerging science — along with a deeper understanding of lipoprotein biology — suggests that not all LDL is created equal. In fact, the very definition of "bad cholesterol" may need an overhaul. The size, density, oxidation state, and metabolic context of LDL particles all influence their atherogenic potential. And importantly, factors like insulin resistance, inflammation, and dietary pattern may play an even bigger role than LDL cholesterol itself in driving heart disease.
In the first two parts of this series, we peeled back the layers of a long-standing dietary narrative. We began with a historical deep dive into how saturated fat became the scapegoat for heart disease — a mix of scientific oversimplification, political momentum, and flawed early data. Then, in part two, we examined what actually happens to saturated fat in the body — tracing its journey from the gut to the liver, and how different dietary contexts (low-carb vs. high-carb, whole vs. ultra-processed) impact triglyceride handling, LDL production, and metabolic health.
In this blog, we get to the heart of the matter — LDL cholesterol itself.
What exactly is LDL? Why do some people have high LDL but no signs of heart disease, while others with “normal” levels still develop atherosclerosis? Is high LDL always dangerous, or does it depend on the type of LDL — and the metabolic context in which it appears?
This is where the conversation shifts from blanket numbers to deeper biology. We'll explore:
What LDL really measures — and the difference between LDL-C, LDL-P, and ApoB
Why particle size (small dense vs. large buoyant) matters
What causes LDL to become truly harmful (hint: it’s not just cholesterol)
Why LDL rises on low-carb, high-fat diets in some people — and what that actually means
How insulin resistance and inflammation often tell us more about cardiovascular risk than LDL levels alone
As we’ll see, LDL isn’t the villain — it’s the context that makes or breaks its impact.
It’s time to cut through the noise and rethink our approach to LDL cholesterol, saturated fat, and cardiovascular health — not based on outdated assumptions, but grounded in physiology, nuance, and modern evidence.
What Exactly Is Cholesterol — and Why Is It So Misunderstood?
Before we dive deeper into LDL — and why not all LDL is created equal — it’s worth taking a step back to understand what cholesterol itself actually is. It's one of the most misunderstood molecules in the health world, and clarifying its role helps reframe the entire conversation around fats, lipoproteins, and heart disease. Let’s take a quick detour.
Cholesterol often gets lumped into the "bad guy" category in health conversations — but this couldn't be further from the truth. At its core, cholesterol is not a fat, nor is it inherently harmful. It’s a waxy, fat-like molecule that plays an essential structural and functional role in the body. In fact, cholesterol is so vital to life that your liver makes most of it — whether you eat it or not. Roughly 80% of the cholesterol in your blood is made endogenously, mainly by the liver, but also by the intestines, adrenal glands, and even cells throughout your body.
Cholesterol is synthesised from a molecule called acetyl-CoA, a key player in energy metabolism. Acetyl-CoA is generated from the breakdown of:
Fats (via beta-oxidation)
Proteins (via amino acid catabolism)
Carbohydrates (via glycolysis and the citric acid cycle)
But here's the kicker: when energy intake exceeds energy demand, especially in the form of refined carbohydrates and sugars, more acetyl-CoA is diverted toward cholesterol and fatty acid synthesis in the liver — a process known as de novo lipogenesis (DNL).
So, even if you’re not eating cholesterol or saturated fat, your liver can still make plenty — and sometimes too much — if it's constantly flooded with excess glucose and insulin. This is part of why a low-fat, high-carb diet isn't always protective against heart disease — especially when it includes lots of ultra-processed foods. The liver responds to this metabolic environment by producing more VLDL, which can increase circulating triglycerides, promote small dense LDL, and ultimately increase atherosclerosis risk — despite low dietary fat or cholesterol intake.
In short: it's not just what you eat — it's how your body is processing what you eat that determines cholesterol dynamics.
Here’s why cholesterol is indispensable:
Cell Membranes: Every single cell in your body relies on cholesterol to maintain membrane fluidity and integrity. Without it, cells would collapse or leak.
Hormone Production: Cholesterol is the precursor to all your steroid hormones, including cortisol, aldosterone, oestrogen, progesterone, and testosterone.
Vitamin D Synthesis: Sunlight converts a cholesterol derivative in your skin into vitamin D, which is crucial for bone health, immunity, and more.
Bile Production: Cholesterol is used by the liver to make bile acids, which are essential for digesting and absorbing dietary fats.

Image Credit: Statinnation
So why the bad reputation?
It comes down to a misunderstanding: when people talk about "cholesterol" on a blood test (like “high cholesterol”), they’re actually referring to cholesterol being transported in the blood by lipoproteins — such as LDL and HDL. But cholesterol itself isn’t inherently good or bad; context is everything.
Problems arise when:
Cholesterol-rich lipoproteins (like LDL) accumulate in the wrong places (like artery walls),
Become oxidised or damaged,
And trigger an inflammatory response that leads to plaque formation.
Even then, cholesterol is not the cause — it's more like the brick that shows up to repair a crack in the wall. The real issue is the underlying inflammation, insulin resistance, or oxidative stress that caused the damage in the first place.
In short: cholesterol isn’t the criminal — it’s often at the scene of the crime because it's trying to help. It’s more like the fireman at the scene of a fire. Just because firemen are always present at fires doesn’t mean they caused the blaze. They’re there to help, to repair, to contain damage. Similarly, cholesterol often shows up at sites of arterial injury and inflammation — not because it’s causing the damage, but because your body is using it to respond and repair. The real arsonists behind the fire? Think chronic inflammation, high blood pressure, insulin resistance, oxidative stress, and smoking — not cholesterol itself.
Blaming cholesterol for heart disease without addressing the root causes is like blaming the fireman for the fire. It's time we shift the focus to what’s igniting the flames in the first place.
What LDL Cholesterol Really Is — and What It’s Not
Now that we’ve cleared up what cholesterol actually is — and why your body works hard to make it — we can shift our focus to how cholesterol travels through the bloodstream. This is where lipoproteins like LDL come in. Let’s begin by clearing up a common misconception: LDL cholesterol is not cholesterol itself — and it’s certainly not a toxin circulating in your blood waiting to clog your arteries.
LDL stands for Low-Density Lipoprotein, which is not a type of cholesterol but a carrier molecule. Think of it as a delivery truck that transports fats (like triglycerides and cholesterol) through the watery environment of your bloodstream. Because fat and water don’t mix well, the body packages fat into lipoproteins so they can travel to where they’re needed — like cell membranes, hormone production, or energy use.
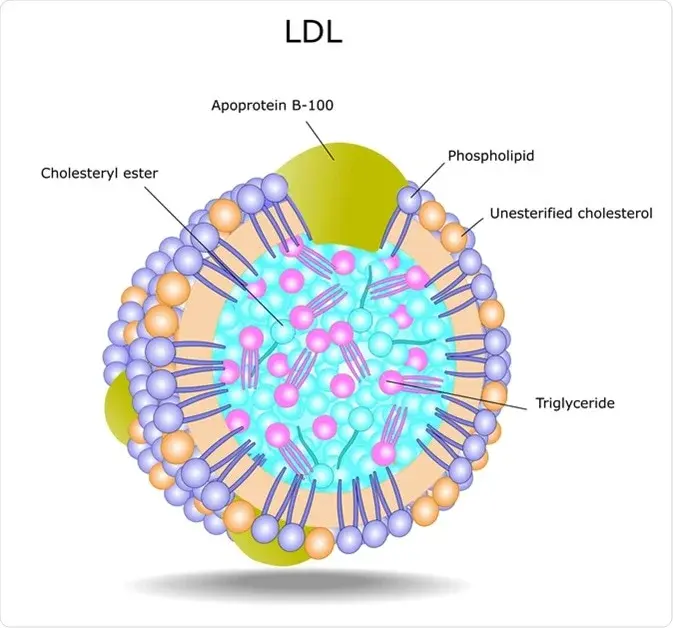
Image Credit: news-medical.net
Here’s how it works biochemically:
VLDL (Very Low-Density Lipoprotein) is produced by the liver to transport triglycerides and some cholesterol to peripheral tissues.
As VLDL circulates, it unloads triglycerides via the enzyme lipoprotein lipase (LPL).
With each offloading step, the lipoprotein becomes smaller and denser, transforming from VLDL → IDL (Intermediate-Density Lipoprotein) → LDL.
LDL now contains mostly cholesterol — which it delivers to cells that need it for membrane repair, hormone synthesis (like testosterone, oestrogen, cortisol), and other essential functions.
So when we measure “LDL cholesterol” (or LDL-C), we’re not measuring the number of LDL particles, but rather the amount of cholesterol being carried within them. It’s a bulk measure, not a count. And that distinction is important, because not all LDL particles carry the same load — and not all are equally risky.
LDL Is Not the Enemy! LDL is a normal, even essential part of human physiology. Every cell in your body relies on cholesterol — and LDL is a key part of how that cholesterol is distributed. It becomes problematic only when:
It’s present in the wrong form (small, dense, oxidised), and
It’s in excess and not cleared efficiently, and
It’s present in a pro-inflammatory, insulin-resistant environment
So instead of asking “How high is your LDL-C?”, the more revealing questions are:
How many LDL particles do you have?
What type of LDL particles are they?
Are they oxidised or inflamed?
What’s your insulin status, metabolic flexibility, and inflammatory load?
This is where modern lipidology starts to challenge conventional wisdom — because elevated LDL-C on its own, especially in metabolically healthy individuals, may not carry the same risk as once assumed.
What LDL Really Measures — And Why That’s Only Part of the Story
When most people talk about their “LDL cholesterol” numbers, they’re referring to LDL-C — the total amount of cholesterol carried within LDL particles. But here’s the problem: LDL-C doesn’t tell you how many particles are carrying that cholesterol, nor what kind of particles they are.
Think of it like this: imagine you're measuring how many people are travelling on a road. LDL-C is like counting the total number of passengers — but not the number of cars. You could have a few large vans, each packed with people, or a traffic jam of small cars, each carrying just one. From a heart disease perspective, it’s the number of cars (LDL particles) that matters more — because more particles on the road means more chances of one breaking down, crashing into the arterial wall, or clogging up the system.
This is where a more nuanced view of LDL becomes essential — and where LDL-P and ApoB come into play. They count the number of vehicles, not just the passengers — and that gives a much clearer picture of risk.
LDL-C: Cholesterol Content
This is the standard measure you’ll see on most lipid panels. It tells you how much cholesterol (in milligrams) is present inside LDL particles, but not how many particles there are or whether those particles are large or small.
You could have two people with identical LDL-C levels, but very different risk profiles:
One may have fewer, larger LDL particles, each carrying more cholesterol (a potentially lower-risk pattern).
The other may have many small LDL particles, each carrying less cholesterol — which increases the chance of those particles slipping into artery walls and triggering inflammation.
LDL-P: Particle Count (number of cars)
LDL-P measures the actual number of LDL particles in the blood, regardless of how much cholesterol each one carries. This is a more direct marker of atherogenic risk, because more particles = more opportunities to penetrate the endothelium (the inner lining of arteries) and kickstart plaque formation.
ApoB: The Essential Identifier
Apolipoprotein B (ApoB) is a protein that sits on the surface of all atherogenic lipoproteins — including LDL, VLDL, IDL, and Lp(a). Since each atherogenic particle carries exactly one ApoB, measuring ApoB gives you an accurate count of the total number of particles capable of contributing to atherosclerosis. Many lipid experts now consider ApoB the most reliable single marker of cardiovascular risk — more so than LDL-C, and in some cases, even more than LDL-P.
Why Particle Size Matters: Small, Dense vs. Large, Buoyant LDL
Not all LDL is created equal. Beyond the number of particles, the size and density of LDL particles can significantly influence cardiovascular risk.
Small, dense LDL (sdLDL): These particles are more likely to penetrate the endothelium, become oxidised, and trigger an inflammatory response — making them more atherogenic. They are commonly elevated in people with insulin resistance, metabolic syndrome, and high triglycerides.
Large, buoyant LDL: These are less likely to slip into arterial walls or become oxidised. They tend to be more common in individuals following low-carb or ketogenic diets, where triglycerides and insulin levels are low.
Here’s the twist: someone on a high-saturated fat, low-carb diet might have elevated LDL-C, but their LDL particles are often larger, and less inflammatory — a very different risk profile from someone with high LDL-C due to metabolic dysfunction.
What Dictates LDL Particle Size?
The size and density of LDL particles are primarily influenced by how lipoproteins are processed in the body, especially in the context of triglyceride metabolism, insulin sensitivity, and overall metabolic health.
Here’s how it works:
1. Triglyceride (TG) Levels and VLDL Metabolism
The liver secretes VLDL particles into the bloodstream — these are rich in triglycerides. As they travel, lipoprotein lipase (LPL) breaks down the TGs, gradually transforming VLDL into IDL, and eventually LDL.
When TG levels are low, VLDL particles are smaller and less TG-rich. The resulting LDL particles are larger and more buoyant.
When TG levels are high (especially in insulin-resistant or high-carb states), the VLDL particles are larger and carry more TGs. These oversized VLDLs lead to the production of smaller, denser LDL particles after TG removal.
So, more TG = more small, dense LDL.
2. Cholesteryl Ester Transfer Protein (CETP) Activity
CETP swaps triglycerides from VLDL with cholesterol esters from LDL and HDL. When VLDL levels are high (as in high-TG states), this exchange leads to:
LDL particles becoming enriched in TGs
TGs are hydrolysed by LPL into free fatty acids (FFAs) and glycerol, which leave the lipoprotein entirely.
Cholesterol esters generally remain within the particle until they are transferred in exchange for TGs via cholesteryl ester transfer protein (CETP), or until the particle is taken up by the liver via receptors.
In high-TG, insulin-resistant states, CETP activity is high → CEs get swapped for TGs in LDL and HDL → these TG-enriched particles are unstable and prone to modification.
This swap is one of the critical steps in creating small dense LDL and lowering HDL cholesterol in metabolic syndrome.
So “shrinking” from cholesterol loss isn’t a major path — the size drop comes primarily from bulk TG removal.
CETP activity is more pronounced in people with:
High insulin
High carb intake (especially refined carbs)
High inflammation
3. Insulin Sensitivity vs. Insulin Resistance
Insulin-sensitive individuals tend to have lower circulating TGs, higher HDL, and less CETP-driven lipid swapping — leading to larger LDL particles.
Insulin-resistant individuals often have higher TGs and lower HDL, setting the stage for small, dense LDL formation.
4. Dietary Composition
Low-carb, higher-fat diets (especially with saturated fats) often lead to larger, buoyant LDL particles, even if LDL-C goes up.
High-carb, low-fat diets — particularly with ultra-processed carbs — tend to increase TGs and promote smaller, denser LDL particles.
Why LDL Particle Size Matters
It is about TG:Cholesterol ratio in the original VLDL. The higher the TG-to-cholesterol ratio in the starting VLDL, the more it will shrink when TGs are removed by LPL.
TG-rich VLDL: When the “fluffy” triglyceride cargo is hydrolysed, the particle loses a lot of volume but keeps its cholesterol and protein skeleton. This makes the resulting LDL smaller in size.
Cholesterol-rich VLDL: Loses less volume because cholesterol is more compact and is not removed in the same way as TGs — so the particle stays larger.
Why not just offload cholesterol to shrink VLDL?
Cholesterol doesn’t leave VLDL in bulk the way TGs do.
TGs are hydrolysed by LPL into free fatty acids (FFAs) and glycerol, which leave the lipoprotein entirely.
Cholesterol esters generally remain within the particle until they are transferred in exchange for TGs via cholesteryl ester transfer protein (CETP), or until the particle is taken up by the liver via receptors.
So “shrinking” from cholesterol loss isn’t a major path — the size drop comes primarily from bulk TG removal.
What dictates the "small" part?
Small = lower particle diameter.This is set by:
Starting particle volume (more TGs = more shrinkage possible)
Degree of TG hydrolysis by LPL
Additional TG removal by hepatic lipase
Extent of particle remodelling via CETP swaps (TG for cholesterol esters)
What dictates the "dense" part?
Dense = mass per unit volume. After TGs (low-density cargo) are removed, the proportion of:
Cholesteryl esters (denser than TGs)
Apolipoprotein B-100 (protein skeleton)
Phospholipid increases relative to particle size.
The particle becomes smaller but keeps much of its heavier cargo → higher density.
Why sdLDL is “dense”
Think of TGs as lightweight packing peanuts and cholesterol esters as heavy bricks:
Start with a big box full of peanuts (TG) and a few bricks (cholesterol).
Remove most of the peanuts.
Now you have a smaller box but still the same bricks → heavier for its size (dense) and more “rigid” in structure.
Why sdLDL is more oxidation-prone
1. Surface composition and size
Smaller particle → higher surface-area-to-volume ratio.This means more of its content is near the surface where it’s exposed to oxidants.
The phospholipid monolayer in sdLDL has often been enriched in polyunsaturated fatty acids (PUFAs) during CETP-mediated swaps and hepatic lipase action. PUFAs are far more oxidation-prone than saturated fats.
Large buoyant LDL has proportionally more monounsaturated/saturated fats in its membrane → more stable.
2. Longer residence time in circulation
SdLDL particles are cleared less efficiently by LDL receptors.
They rely more on scavenger receptor pathways for clearance, which are slower.
Longer time in plasma = more exposure to reactive oxygen species (ROS) and glycation agents.
3. Electrical charge and binding to arterial proteoglycans
SdLDL tends to have changes in apoB conformation that make it more negatively charged.
This increases its binding affinity for arterial wall proteoglycans → particles stick around in the subendothelial space longer → higher oxidation risk inside the artery wall.
4. Higher association with systemic oxidative stress
SdLDL is more common in insulin resistance, high TG states, and metabolic syndrome.
These states already come with higher ROS production and lower antioxidant capacity, making oxidation more likely.
Analogy
Think of LDL particles as delivery vans carrying the same boxes of cholesterol:
Large buoyant LDL = big, slow-moving vans with thick steel panels → harder to damage, shorter routes.
Small dense LDL = smaller vans with thinner panels, more fragile materials on the outside, and they’re stuck in traffic longer → more dents and rust.
In Summary:
LDL particle size is a reflection of the metabolic environment:
High TGs + high CETP activity → small, dense LDL
Low TGs + low CETP activity → large, buoyant LDL
So, while LDL-C just measures the cholesterol carried, LDL particle size and number tell us much more about actual cardiovascular risk.
When Is High LDL More Concerning vs. Less Concerning?
Context | LDL Profile | Other Markers | Cardiovascular Risk |
Metabolically Unhealthy (e.g. insulin resistance, metabolic syndrome, chronic inflammation, high TGs, low HDL) | Often small, dense LDL | High ApoB, high triglycerides, low HDL, elevated CRP | High risk — LDL more likely to be oxidised and promote plaque formation |
Hypercaloric, High-Carb, Ultra-Processed Diet | Increased VLDL → small, dense LDL | Elevated insulin, TGs, possibly high LDL-P/ApoB | Moderate to high risk, especially with systemic inflammation |
Low-Carb, High-Saturated Fat Diet (e.g. ketogenic or carnivore) | Often large, buoyant LDL | Low TGs, high HDL, low inflammation, low CRP, normal ApoB | Low risk — especially in active, insulin-sensitive individuals |
Lean Mass Hyper-Responder (LMHR) | Very high LDL-C, but large particles | Low TGs, high HDL, low ApoB | Still debated, but emerging data suggest low actual risk in this unique phenotype |
Inflammation-Free, Whole-Food Diet (e.g. low-processed carbs) | Mixed LDL pattern | Balanced TGs/HDL, low CRP | Likely low to moderate risk, depending on ApoB and insulin sensitivity |
Key Takeaway: It’s not just how high your LDL-C is — it’s how many particles are circulating (ApoB), what type they are (particle size and density), and what environment they’re in (inflammatory vs. insulin-sensitive).
What Causes LDL to Become Truly Harmful?
By now, we’ve seen that not all LDL is created equal. But the real turning point — when LDL goes from a passive transporter to a genuine cardiovascular threat — depends heavily on the metabolic environment it’s circulating in. Elevated LDL on its own is not necessarily harmful, especially if the particles are large, buoyant, and not oxidised. The real problem arises when LDL gets oxidised, inflamed, or trapped in arterial walls — and this tends to happen under specific metabolic conditions.
Here’s what truly pushes LDL into dangerous territory - Arsonists
1. Insulin Resistance
When insulin sensitivity drops — typically due to a diet high in ultra-processed carbs, lack of activity, and chronic overeating — it sets off a cascade:
The liver pumps out more VLDL particles (rich in triglycerides).
These VLDLs exchange contents with LDL particles, creating more small, dense LDL.
Insulin resistance contributes to endothelial dysfunction by impairing the ability of blood vessels to produce nitric oxide (NO) — a molecule essential for keeping arteries flexible and dilated. Without enough NO, the endothelium (the inner lining of blood vessels) becomes more permeable, inflamed, and adhesive. This makes it easier for LDL particles — especially small, dense, oxidised ones — to slip into the arterial wall, become trapped, and trigger an immune response. Over time, this process lays the groundwork for plaque formation and atherosclerosis.
2. Chronic Inflammation
Inflammation is the match that lights the fire. Once LDL particles become oxidised (especially small dense ones) inside the intima, they can trigger immune cells to attack them, creating foam cells and initiating plaque formation. High-sensitivity CRP (hs-CRP) is one marker that reflects this low-grade, chronic inflammation.
Without inflammation, many LDL particles — even if elevated — may never become problematic. Just as the fireman showing up at the scene of a fire didn’t cause the fire — he’s there trying to help. But if fires are constantly breaking out (due to inflammation, oxidative stress, high blood sugar, endothelial damage), then seeing firemen everywhere might lead you to blame them. In reality, the arsonists are things like chronic inflammation, insulin resistance, and oxidative damage — not LDL itself.
When the metabolic environment is calm — low inflammation, low oxidative stress, good insulin sensitivity — LDL particles circulate, deliver nutrients, and return to the liver without incident. But in an inflamed environment, those same particles can get trapped, oxidised, and turned into plaque.
3. High Triglycerides and Low HDL
This classic triad — high TG, low HDL, and insulin resistance — is a hallmark of metabolic syndrome. It’s strongly linked to:
A higher number of small, dense LDL particles
Increased ApoB levels
Greater cardiovascular risk
In contrast, people with low TG and high HDL often have fewer and larger LDL particles — which are far less atherogenic.
4. Oxidative Stress
LDL becomes especially dangerous when it’s oxidised — damaged by free radicals. This makes it sticky, inflammatory, and far more likely to initiate atherosclerotic plaques. Smoking, poor diet, pollution, and stress can all drive oxidative stress.
5. Glycation
Elevated blood sugar (often from excess refined carbs) leads to glycation of LDL particles, making them more likely to become oxidised and taken up by immune cells in artery walls. This is especially concerning in people with type 2 diabetes or prediabetes.
The Bottom Line
LDL cholesterol doesn’t operate in isolation. It becomes dangerous not because it’s inherently bad, but because of the metabolic environment it exists in. If you're inflamed, insulin-resistant, and eating a diet rich in ultra-processed foods, even normal LDL levels can be a problem. But in someone who's metabolically healthy — with low insulin, low inflammation, low triglycerides, and good HDL — even higher LDL-C may pose minimal actual risk, especially if ApoB levels remain moderate and LDL particles are large.
LDL in a Healthy vs. Unhealthy Metabolic State
Factor | Healthy Metabolic State | Unhealthy Metabolic State |
Insulin Sensitivity | High | Low (insulin resistance) |
Inflammation | Low | Chronic, low-grade inflammation |
Triglycerides (TGs) | Low | Elevated |
HDL Cholesterol | High | Low |
LDL Particle Size | Large, buoyant LDL (less atherogenic) | Small, dense LDL (more atherogenic) |
LDL Particle Number (LDL-P) | Normal to slightly elevated | Elevated (often with normal or low LDL-C) |
ApoB Levels | Normal/Elevated (*HFD) | Elevated |
Endothelial Function | Healthy, resilient | Dysfunctional (easier LDL entry into arteries) |
Oxidative Stress | Low | High (promotes LDL oxidation) |
LDL Oxidation | Unlikely | Likely |
Plaque Formation Risk | Low | High |
*HFD: People on high fat diets can see high ApoB counts due to increased cargo. If TG:HDL levels are under 1, Remnant Cholesterol is under 0.6, CRP levels are low; for most part the high ApoB count could be harmless!
Summary Box:
LDL itself isn’t the root cause. In a healthy metabolic state, it does its job and moves on. In a damaged, inflamed environment, it becomes vulnerable, oxidised, and trapped — setting the stage for atherosclerosis.
Rethinking Cholesterol Guidelines and Risk Assessment
Now that we've explored the nuance behind LDL — what it really is, what drives it to become harmful, and how metabolic health reshapes its impact — it's time to step back and ask: Are our current cholesterol guidelines giving us the full picture?
For decades, standard risk assessments have focused on total cholesterol and LDL-C (the amount of cholesterol inside LDL particles) as primary markers of cardiovascular risk. But as we’ve seen, LDL-C doesn’t reveal how many particles are circulating, what type they are, or whether the metabolic environment encourages those particles to cause damage.
This means someone with "high LDL-C" but:
low inflammation,
excellent insulin sensitivity,
low triglycerides, and
high HDL
…might be at far lower cardiovascular risk than someone with normal LDL-C but:
high triglycerides,
low HDL,
insulin resistance, and
chronic inflammation.
Yet both would likely be treated very differently under current clinical guidelines.
It's becoming increasingly clear that context matters — and risk should be evaluated using a more comprehensive lens that includes:
ApoB (the number of atherogenic particles),
LDL particle size and number,
HDL levels and function,
triglycerides,
fasting insulin or HOMA-IR, and
markers of inflammation like hs-CRP.
In the next blog, we’ll explore the pathophysiology of Arteriosclerosis, what a more modern, metabolically informed approach to cardiovascular risk assessment might look like — and how it could shift both clinical practice and personal health decisions.
📢 A Note on "Living Science"
Science is not a static destination; it is a moving target. While the principles of Turnover, Signaling, and Tension are grounded in decades of metabolic research, new peer-reviewed data emerges every day.
I am committed to accuracy. If you are a researcher, clinician, or dedicated student of physiology and you find a piece of data here that does not align with the latest high-quality evidence, please reach out. I welcome civil, evidence-based corrections. My goal is to keep this resource as the most accurate "No-Nonsense" guide to protein on the internet. Let’s get better together.
*Disclaimer:
The information provided in this blog is for educational and informational purposes only and should not be construed as medical advice. While every effort is made to ensure accuracy, the content is not intended to replace professional medical consultation, diagnosis, or treatment. Always seek the guidance of a qualified healthcare provider with any questions regarding your health, medical conditions, or treatment options.
The author is not responsible for any health consequences that may result from following the information provided. Any lifestyle, dietary, or medical decisions should be made in consultation with a licensed medical professional.
If you have a medical emergency, please contact a healthcare provider or call emergency services immediately.



Comments