


How and Why Seed Oils and Plant Sterols Contribute to Artherosclerosis
- S A

- Oct 14, 2025
- 15 min read
Updated: Feb 19
For decades, saturated fat and cholesterol have been portrayed as the primary culprits in heart disease. But when we examine the pathophysiology of atherosclerosis through the lens of clotting, oxidative stress, and red blood cell (RBC) health, a very different story emerges. Seed oils rich in polyunsaturated fatty acids (PUFAs) and plant sterols appear to play a far more destabilising role than natural saturated fats or dietary cholesterol.
What Exactly Are PUFAs and Plant Sterols?
Polyunsaturated Fatty Acids (PUFAs)
Definition: Fatty acids with more than one double bond in their carbon chain. These double bonds make the molecule more fluid but also more chemically unstable.
Types:
Omega-6 PUFAs (e.g. linoleic acid) – abundant in seed oils and modern processed food.
Omega-3 PUFAs (e.g. EPA, DHA, ALA) – found in fish, flax, and some plants; anti-inflammatory when balanced with omega-6.
Problem: High omega-6 intake (typical modern ratio 15:1 vs ancestral 1–3:1) drives pro-inflammatory pathways, endothelial stress, and promotes oxidation of lipoproteins and membranes.
Plant Sterols (Phytosterols)
Definition: Plant-derived molecules that look almost identical to cholesterol, except for small side-chain differences.
Role in Plants: Structural component of plant cell membranes.
Plant Sterols → Membrane Instability and Cholesterol Crystals
Plant sterols (phytosterols) resemble cholesterol but don’t integrate smoothly into cell membranes. When absorbed and incorporated into RBCs, they cause structural mismatches:
Membranes become patchy and fragile, leading to premature haemolysis and elevated turnover (more reticulocytes, more microparticles).
Fragile RBCs trapped in clots release sterol- and cholesterol-rich debris that seeds plaque growth.
Within arterial lesions, sterol-laden debris crystallises, piercing tissues like tiny needles, amplifying inflammation and promoting rupture.
Role in Humans:
Poorly absorbed in the gut (most is excreted).
When absorbed, they can insert into red blood cell (RBC) membranes and arterial plaques, where their imperfect “fit” destabilises membranes.
They also crystallise more easily than cholesterol, creating mechanical stress within arterial plaques.
Medical Context: Added to margarines and “heart healthy” foods to lower LDL cholesterol — but this effect does not necessarily translate into better outcomes, and may carry hidden risks.
How Seed Oils Are Made (vs. Natural Fats)
Natural fats like butter, ghee, olive oil, or coconut oil are typically extracted with minimal processing (churning, pressing, or gentle heating).
Seed oils, by contrast, require intensive industrial methods:
Collection & cleaning of seeds (soy, corn, sunflower, cottonseed, etc.).
Mechanical pressing to extract some oil.
Solvent extraction (usually hexane) to maximise yield.
Refining to remove unwanted particles.
Bleaching to remove colour.
Deodorising at very high heat, which can generate oxidised PUFA byproducts and trans fats.
End product: a clear, shelf-stable oil marketed as “vegetable oil,” but structurally fragile due to high PUFA content and already containing oxidised lipids.
Why This Matters
PUFAs = chemically unstable, prone to oxidation in the bottle, frying pan, and bloodstream.
Plant sterols = cholesterol mimics that sneak into human membranes, making RBCs and plaques fragile.
Seed oils = industrial products that deliver both PUFAs and plant sterols in high amounts, a modern dietary exposure far outside ancestral norms.

Image Credit: Oilmachinecn
How and Why Seed Oils and Plant Sterols Contribute to Atherosclerosis
Now that we’ve unpacked what PUFAs and plant sterols actually are — and how seed oils deliver them in large amounts — the next question is: what do they do inside the human body?
This is where physiology connects to pathology. On paper, polyunsaturated fats and plant sterols may sound harmless, even “heart healthy.” In reality, their chemistry sets the stage for vascular damage
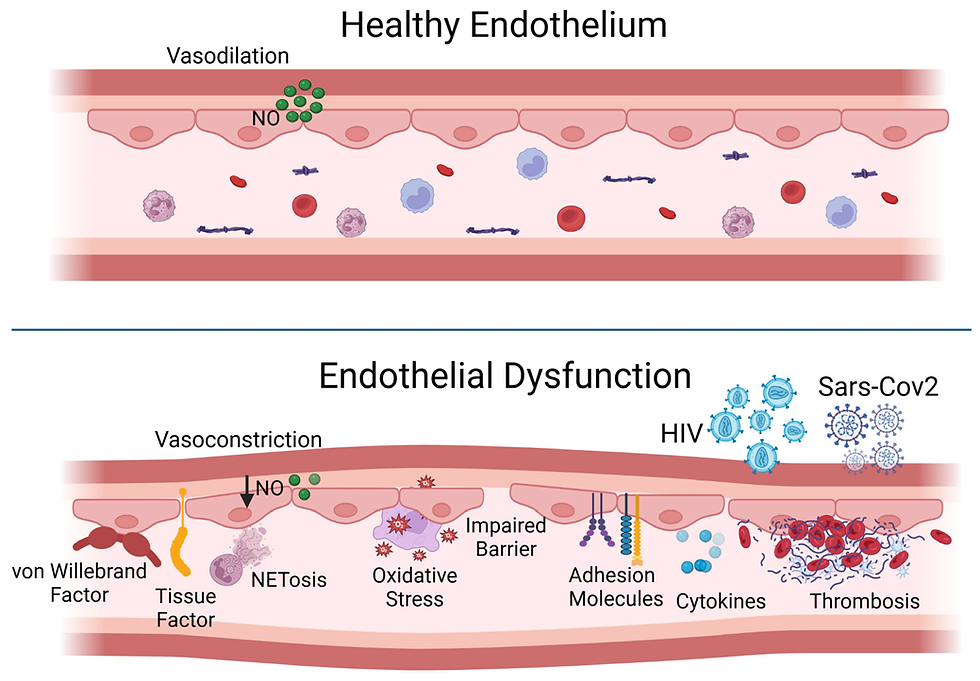
PUFAs are chemically unstable; their multiple double bonds make them prone to oxidation. Once oxidised, they generate free radicals, damage red blood cells and lipoproteins, and inflame the endothelium.
Plant sterols look enough like cholesterol to sneak into cell membranes and arterial plaques, but their “imperfect fit” destabilises both red blood cells and the plaque structure itself. They also crystallise more easily, creating mechanical stress that can trigger rupture.
Seed oils concentrate both of these problematic molecules — delivering an unprecedented, industrial dose of fragile fats and sterols that our biology never evolved to handle.
The result? A perfect storm of endothelial damage, clot formation, and plaque instability that shifts the cardiovascular risk landscape. This is not the slow, stable narrowing of arteries by cholesterol-rich fatty streaks; it’s the setup for sudden, catastrophic blockage driven by clotting and rupture.
Red Blood Cells → Clotting
Red blood cells are not passive oxygen carriers — they actively shape the clotting environment. Their membranes are normally stabilised by cholesterol, which gives them the right balance of flexibility and resilience to squeeze through tiny capillaries.
When membranes are disrupted, RBCs lose deformability and become fragile.
Fragile cells rupture prematurely, releasing pro-thrombotic microparticles, haemoglobin, iron, and membrane fragments rich in cholesterol and lipids.
Rigid, sticky cells sludge in microcirculation, pile into fibrin networks, and make clots denser and harder to dissolve.
Healthy RBCs = smooth flow and stability. Damaged RBCs = fertile ground for clot formation and plaque instability.
Let’s expand the connection between RBCs, fragility/lifespan, and clotting.
RBCs as key players in thrombosis
RBC membranes are rich in cholesterol and phospholipids. Their composition (influenced by diet, sterols, seed oils, oxidative stress) affects flexibility, charge, and survival.
Damaged or fragile RBCs release contents such as free haemoglobin, phosphatidylserine, and microparticles. These are highly pro-thrombotic because they activate the clotting cascade and provide surfaces for thrombin generation.
RBCs are major structural components of clots. They become trapped in fibrin meshes and determine clot size, density, and oxygen-trapping capacity. “Red” thrombi (fibrin + RBC rich) are particularly dangerous in coronary and venous events.
Plant sterols, seed oils, and fragility
Phytosterols can incorporate into RBC membranes, destabilising them, making cells more fragile and prone to haemolysis.
Oxidised PUFA (seed oils) in membranes increases susceptibility to peroxidation. Peroxidised membranes fragment more easily, leading to microparticles.
Result: higher turnover, higher reticulocyte count, and pro-thrombotic microparticles in circulation.
RBC lifespan and HbA1c paradox
Short-lived RBCs (fragile): spend less time exposed to glucose → HbA1c falsely low, but clotting risk high (because of ongoing RBC breakdown, microparticle release, and oxidative stress).
Long-lived RBCs (stable): HbA1c may rise (artificially, because cells live longer and accumulate more glycation), but clotting risk may be lower because RBCs are resilient, deformable, and less prone to rupture.
This explains why HbA1c can be misleading in metabolic health assessments unless paired with glucose monitoring or reticulocyte counts.
RBC deformability and blood flow
Healthy RBCs are flexible, squeezing through tiny capillaries and supporting smooth flow.
In insulin resistance, high TG:HDL ratio, and oxidative stress, RBCs lose deformability, become “sticky,” and promote sludging in microcirculation.
Stiffer RBCs amplify shear stress on endothelium, increasing risk of plaque rupture and subsequent clot formation.
RBCs inside clots
Clots in coronary thrombosis are often RBC-rich with cholesterol trapped inside. This cholesterol largely comes from RBC membranes, not just lipoproteins.
Over time, trapped RBCs break down inside the clot, leaving behind cholesterol crystals and iron, which further irritate the plaque and perpetuate inflammation and calcification.
This links the “clotting theory” of atherosclerosis to the RBC fragility/membrane composition hypothesis.
Practical markers and implications
Reticulocyte count: high = lots of RBC turnover = oxidative stress/fragility.
HbA1c vs CGM mismatch: can reveal RBC lifespan changes.
RBC deformability tests (e.g. ektacytometry, though rarely used clinically) show hidden risks.
LDH (lactate dehydrogenase) and haptoglobin levels can also reflect haemolysis burden.
Pulling it together
High TG:HDL ratio → insulin resistance, oxidative stress → fragile, sticky RBCs + pro-thrombotic plasma.
Fragile RBCs (via plant sterols, seed oils, oxidative damage) → more microparticles, more cholesterol debris inside clots.
Long-lived RBCs (e.g. in healthier diets) → HbA1c may look worse, but actual clotting risk is lower because RBCs are stable and non-thrombogenic.
So RBC health is a missing bridge between metabolism, lipids, and the clotting model of heart disease. They act as both carriers of oxygen and lipids, and as co-conspirators in the final event of arterial occlusion.
Technical section: Can be skipped
The “stickiness” and loss of deformability of RBCs comes from both mechanical/structural changes in their membranes and biochemical signalling changes in their environment. Let’s break it down:
Biophysics of cell membranes
1. Normal RBC membrane composition
The red cell membrane is a lipid bilayer made mostly of phospholipids + cholesterol.
Cholesterol is not just “filler”: it modulates fluidity, stability, and deformability of the membrane.
In RBCs, cholesterol is especially important because they must squeeze through capillaries narrower than themselves without breaking. Too much rigidity or too much fluidity = fragility.
2. Normal deformability = survival mechanism
A healthy RBC is basically a bag of haemoglobin in a flexible lipid–protein shell.
Cholesterol and specific membrane proteins (like spectrin, ankyrin, band 3) give it both strength and flexibility.
This deformability is critical: capillaries are often smaller in diameter than the RBC itself (3–4 μm capillary vs 7–8 μm RBC). A normal RBC must elongate and squeeze through.
3. Why RBCs lose deformability
Several insults change the membrane and cytoskeleton:
Oxidative stress: Reactive oxygen species damage membrane lipids and proteins (especially spectrin and band 3). This makes the RBC rigid and prone to fragmentation.
Glycation (diabetes/insulin resistance): Non-enzymatic glycation stiffens membrane proteins, reducing elasticity.
Abnormal lipid composition:
PUFA peroxidation → weak membranes, cross-linking.
Phytosterol incorporation → microdomain instability.
Excess cholesterol loading → too rigid.
ATP depletion: RBCs rely on ATP-driven ion pumps to maintain volume and flexibility. In metabolic syndrome, ATP is often depleted, leading to dehydrated, stiff RBCs.
Ageing: As RBCs age, they lose membrane area via vesiculation, reducing their surface-to-volume ratio → less deformability.
4. Why RBCs become “sticky”
Phosphatidylserine (PS) exposure: In stressed/damaged RBCs, PS flips from the inner leaflet of the membrane to the outer surface. This is a strong pro-coagulant signal, making RBCs bind to endothelium and platelets.
Loss of glycocalyx (surface coat): Normally RBCs carry a negatively charged glycocalyx that repels other cells. Damage reduces this charge, making them clump.
Microparticle shedding: Fragile RBCs release vesicles rich in PS and oxidised phospholipids → these act as clotting “hotspots.”
Endothelial adhesion: In inflammatory states, endothelial cells express adhesion molecules (VCAM-1, ICAM-1, P-selectin), and sticky RBCs latch on.
5. How this leads to “sludging”
Rigid RBCs cannot squeeze smoothly through capillaries → they pile up, obstructing flow.
Stickier surfaces → RBC–RBC and RBC–platelet aggregates.
Reduced nitric oxide (NO) bioavailability (common in insulin resistance, oxidative stress) → RBCs can’t release NO to help vessels dilate, worsening microvascular congestion.
This microcirculatory “sludge” → local hypoxia, endothelial damage, and further pro-thrombotic signalling.
6. Connection to clotting and heart disease
Rigid, sticky RBCs not only worsen microcirculation but also amplify clot formation:
They get trapped more easily in fibrin meshes.
They make clots denser, less permeable, and harder to dissolve.
Their contents (cholesterol, haemoglobin, iron) promote inflammation and plaque instability when they rupture inside thrombi.
7. How phytosterols differ from cholesterol
Phytosterols (plant sterols) look chemically similar to cholesterol, but they have extra side chains (usually an ethyl or methyl group).
These side chains make them pack less neatly into lipid bilayers compared to cholesterol.
Result: instead of providing the perfect “fit” that cholesterol does, phytosterols create membrane irregularities and microdomains of instability.
8. Consequences for RBCs
Reduced membrane flexibility: RBCs need optimal fluidity to deform. Phytosterol substitution disrupts the delicate balance, making membranes stiffer or patchy.
Increased permeability and fragility: The bilayer becomes more prone to rupture under mechanical stress (e.g., squeezing through small vessels).
Oxidative vulnerability: Phytosterol-containing membranes may be more sensitive to lipid peroxidation, further weakening them.
Haemolysis & microparticle release: Fragile RBCs lyse earlier, spilling haemoglobin and releasing pro-thrombotic membrane fragments.
9. Supporting evidence
In sitosterolaemia (a rare condition where people absorb plant sterols excessively), patients develop fragile RBCs and haemolytic anaemia. This is a strong real-world example that phytosterol incorporation into RBCs destabilises them.
Experimental studies show phytosterols alter membrane order and elasticity compared to cholesterol, consistent with the above.
10. Why this matters for clotting
Fragile RBCs = more turnover, more reticulocytes, and more microparticles.
RBC microparticles provide surfaces for clotting enzymes and promote thrombin generation.
Bursting RBCs inside plaques seed cholesterol crystals, which act like needles inside clots, worsening inflammation and instability.
So in short: RBCs lose deformability when their membranes or cytoskeleton are damaged (oxidation, glycation, bad lipids, ATP depletion). They become sticky when they expose pro-coagulant signals (like phosphatidylserine) and lose their natural repulsive charge. Together, this causes sludging in capillaries and contributes directly to thrombosis.
Bringing It Together
The emerging picture is that RBC fragility, oxidative stress, sterol-induced membrane instability, and omega-6 imbalance feed directly into the clotting model of atherosclerosis. Seed oils and plant sterols compromise RBC health, accelerate haemolysis, and produce more thrombogenic debris than saturated fat or cholesterol ever could.
The “villain” may not be the saturated fat that strengthens membranes, but rather the unstable PUFAs, excessive omega-6, and unnatural sterols that weaken them and tilt the system toward clotting and chronic vascular injury.
PUFA → Oxidation and Rigidity
PUFAs are structurally unstable because of their multiple double bonds. When incorporated into RBC membranes or lipoproteins, they are highly prone to oxidation.
Lipid peroxidation products (like malondialdehyde and 4-HNE) crosslink membrane proteins, stiffening the RBC shell.
Oxidised RBCs lose deformability, stick to vessel walls, and shed microparticles that accelerate coagulation.
Inside clots and plaques, oxidised PUFA residues perpetuate inflammation, make fibrin networks more resistant to breakdown, and promote calcification.
This oxidative fragility makes PUFA-rich seed oils a powerful driver of both clot initiation and clot persistence.

Image Credit: Oilrefineryplant
Omega-6 Excess → Inflammation and Imbalance
Omega-6 fatty acids (especially linoleic acid) are essential — the body needs them for cell membranes and signalling molecules. But excess omega-6 intake from seed oils disrupts the natural omega-6 to omega-3 ratio, shifting physiology toward a pro-inflammatory, pro-thrombotic state.
Omega-6 is converted into arachidonic acid, which feeds into the production of inflammatory eicosanoids (prostaglandins, thromboxanes, leukotrienes).
These molecules heighten platelet aggregation, vasoconstriction, and clotting tendency.
Meanwhile, omega-3 (EPA/DHA) normally balances this by producing anti-inflammatory, anti-thrombotic mediators (resolvins, protectins).
A modern Western diet often pushes the ratio from a traditional ~1:1–2:1 balance to 15:1 or higher, creating a systemic tilt toward inflammation and clot formation.
Thus, it isn’t that omega-6 is “bad” — it’s the imbalance driven by seed oils that primes the vascular system for chronic inflammation and acute clotting events.
Why are PUFAs more prone to oxidation?
Polyunsaturated fatty acids (PUFAs) have multiple double bonds in their carbon chain. Each double bond is a weak point where hydrogen atoms can be stripped away by reactive oxygen species (ROS). This process — lipid peroxidation — is like a chain reaction fire:
Initiation – ROS (from metabolism, smoking, pollution, excess glucose, etc.) attack a double bond, stealing a hydrogen.
Propagation – The unstable PUFA radical reacts with oxygen, forming lipid peroxides. These then attack neighbouring PUFAs in membranes and lipoproteins, spreading the damage.
Termination – Crosslinked or fragmented products (e.g. malondialdehyde, 4-HNE) accumulate, stiffening membranes and damaging proteins/DNA.
Because saturated fats lack double bonds, and monounsaturated fats have only one, they are far more resistant to oxidation. PUFAs are uniquely fragile.
What happens when PUFAs oxidise?
RBC membranes stiffen – Oxidised PUFAs form rigid, crosslinked structures in the red blood cell (RBC) membrane. This reduces deformability, making cells “sticky” and prone to sludging in capillaries, increasing clot risk.
LDL particles oxidise (oxLDL) – PUFA-rich LDL is unstable. Once oxidised, oxLDL is engulfed by macrophages, forming foam cells and initiating plaque.
Inflammation is triggered – Oxidised PUFA products (like 4-HNE) act as direct signalling molecules, upregulating adhesion molecules on the endothelium and recruiting immune cells to the vessel wall.
Coagulation is amplified – Damaged membranes release procoagulant signals (e.g. phosphatidylserine exposure on RBCs and platelets), promoting clot initiation.
From oxidation to atherosclerosis
Step 1: Endothelial damage – Oxidised lipids irritate and inflame the inner lining of arteries.
Step 2: Sticky blood – Rigid RBCs and activated platelets increase the chance of sludging and microclots.
Step 3: Plaque growth – Macrophages ingest oxLDL, forming foam cells and fatty streaks.
Step 4: Instability – PUFA-rich membranes and sterol inclusions crystallise or oxidise, making plaques more fragile and rupture-prone.
Step 5: Heart attack or stroke – The sudden rupture of a fragile plaque triggers a large clot that blocks the artery.
The paradox of “heart healthy” PUFAs
While PUFAs may lower LDL-C levels in the bloodstream, their instability means they generate more oxidised LDL, a far stronger predictor of cardiovascular events. In other words, you may lower your cholesterol number but worsen the quality of your lipids and blood cells.
Stage | Saturated fats / dietary cholesterol | PUFA (seed oils) / plant sterols |
Initial endothelial stress / inflammation | Less prone to oxidation; in whole-food contexts tends not to generate reactive lipid aldehydes that damage endothelium. May contribute to metabolic signals in excess, but not via easy peroxidation. | High PUFA intake increases substrate for lipid peroxidation; omega-6 excess feeds pro-inflammatory eicosanoids. Seed oils therefore raise oxidative and inflammatory burden on the endothelium. |
RBC membrane composition & deformability | Cholesterol fits well into membranes and helps preserve optimal fluidity and deformability; RBCs remain flexible and less likely to rupture. | PUFA incorporation increases peroxidation susceptibility; plant sterols insert poorly and create packing defects. Both pathways reduce deformability, increase fragility or rigidity, and promote microparticle release. |
Lipoprotein behaviour (VLDL/LDL/HDL) | Saturated fats tend to raise LDL in some contexts but produce particles that are less oxidisable. Dietary cholesterol largely integrates into regulated pools; HDL function preserved in low-processed diets. | PUFA-rich lipoproteins are more susceptible to oxidation; high omega-6 shifts to pro-thrombotic eicosanoid signalling. Plant sterols lower LDL-C numerically (by blocking absorption) but increase sterol content in tissues and RBCs. |
Plaque formation & composition | Lipid deposition occurs but membranes and lipids are less oxidised; plaques may develop slowly and often provoke collateral formation. | Oxidised lipids and ruptured RBCs seed plaques with peroxidised residues and sterol-rich debris. Higher tendency for cholesterol/sterol crystallisation and inflammatory, unstable lesions. |
Crystal formation & mechanical destabilisation | Less tendency to precipitate sharp crystals at equivalent loads because cholesterol integrates better into lipid phases. | Plant sterols have lower solubility in lipid matrices and crystallise more readily; oxidative damage reduces solvation capacity — crystals form earlier and act as mechanical irritants that provoke rupture. |
Clotting / thrombus characteristics | Clots form when plaques rupture, but blood may be less primed for extreme thrombosis if RBCs and plasma are less pro-thrombotic; fibrin nets less enriched with oxidised debris. | Damaged/fragile RBCs, PS exposure, microparticles and oxidised lipids catalyse thrombin generation. Clots are denser, more RBC-rich and harder to lyse — driving sudden occlusions (MI/stroke). |
Clinical pattern / outcome | Gradual stenosis more common; compensatory collateralisation can mitigate acute occlusion in many cases. | Greater propensity for sudden arterial occlusion from thrombosis; higher risk of abrupt MI/stroke despite similar or lower LDL-C readings. |
Net mechanistic implication | Supports membrane stability and resistance to oxidation (in whole-food contexts); surrogate LDL changes need physiological context. | Promotes oxidation, membrane instability and sterol crystallisation — mechanistically coherent with a clot-centric model of CVD even when LDL-C falls. |
Why Are PUFA and Plant Sterols Still Promoted as ‘Heart Healthy’?
At this point, it’s worth asking: if polyunsaturated seed oils and plant sterols disrupt red blood cells, fuel oxidation, promote inflammation, and crystallise in plaques — then why are they still recommended as “cardioprotective”? The answer lies not in physiology, but in the history of nutrition science and the reliance on surrogate markers.
The Cholesterol Hypothesis Legacy
For decades, the dominant model has been: “lower LDL cholesterol = lower heart disease risk.”
PUFA oils and plant sterols do indeed lower measured LDL-C in blood tests. Sterols displace cholesterol from intestinal absorption, and PUFA oils upregulate LDL receptor activity.
But lowering LDL-C on paper does not necessarily mean lowering actual cardiovascular events, especially if the mechanism destabilises vessels in other ways (oxidation, fragility, crystallisation).
Short-Term vs Long-Term Outcomes
Clinical trials often measure short-term changes in LDL-C, triglycerides, or total cholesterol rather than hard outcomes like myocardial infarction or all-cause mortality.
This makes interventions that tweak lab values look “beneficial,” even if long-term physiology tells another story.
The Saturated Fat Scapegoat
Since the 1960s, saturated fat has been demonised as the driver of heart disease. Once that narrative took hold, alternatives like seed oils and sterols were marketed as the “heart-friendly” replacements — without rigorous mechanistic vetting.
The food industry had a vested interest in cheap, industrial seed oils, making them the default substitute for butter, lard, and tallow.
Regulatory Blind Spots
Plant sterols are approved for cholesterol-lowering spreads and supplements because they consistently reduce LDL-C in trials.
Yet rare conditions like sitosterolaemia show us exactly what happens when plant sterols accumulate: premature atherosclerosis. Despite this, the focus has stayed narrowly on LDL reduction rather than sterol crystallisation or vascular damage.
Omega-6:3 Ratio Ignored
Omega-6 linoleic acid is essential in small amounts, but when consumed in massive excess (relative to omega-3s), it shifts the body toward a pro-inflammatory state.
Instead of addressing balance, guidelines promote “more PUFA” without context — effectively pushing a skewed 20:1 ratio that no ancestral diet ever contained.
Foods Rich in PUFA and Plant Sterols
Category | Common Foods | Notes |
High in Omega-6 PUFAs (seed oils & processed foods) | Soybean oil, corn oil, sunflower oil, safflower oil, cottonseed oil, grapeseed oil | Widely used in processed foods, salad dressings, baked goods, fried foods; highly oxidisable. |
Margarine, vegetable shortening | Industrially hydrogenated/processed; can also contain trans fats. | |
Packaged snacks, biscuits, crackers, chips, fast food | Often fried in or made with PUFA-rich seed oils. | |
Poultry skin, grain-fed pork, farmed fish (esp. tilapia, catfish) | Higher omega-6 when animals are grain-fed vs grass-fed/wild. | |
High in Plant Sterols (added or naturally occurring) | Fortified foods: “cholesterol-lowering” margarines (e.g. Benecol, Flora Pro-Activ), sterol-enriched yoghurt drinks | Often marketed as heart-healthy for lowering LDL-C. |
Nuts & seeds: almonds, walnuts, sunflower seeds, sesame, pumpkin seeds | Natural sterol content, though effect depends on intake. | |
Legumes: soybeans, peas, chickpeas | Soy oil and soy products are major sterol sources. | |
Whole grains: wheat germ, rye, rice bran | Wheat germ oil is especially high in sterols. | |
Vegetable oils (again): corn oil, soybean oil, canola oil | Dual role — high in both omega-6 PUFAs and plant sterols. |
Key physiological context
Omega-6 PUFAs are essential in small amounts, but modern diets push the omega-6:3 ratio to 15:1 or higher (ancestrally closer to 1–3:1), driving pro-inflammatory signalling.
Plant sterols lower LDL-C on a lab report but can incorporate into RBC and arterial membranes, destabilise them, and crystallise more easily than cholesterol.
Processed food overlap: Many “cholesterol-lowering” or “vegetable oil–based” products deliver both high omega-6 PUFA and plant sterols in one package.
Critical Thinking: Surrogate Success vs Biological Reality
Surrogate success: LDL-C reduction makes PUFA and plant sterols look good on a lab report.
Biological reality: These compounds can destabilise membranes, fuel oxidation, and crystallise inside plaques.
A truly heart-healthy fat should support cell membrane stability, resist oxidation, and integrate smoothly into physiology — qualities more consistent with monounsaturated fats (olive oil) or even saturated fats in natural whole foods, than with industrial seed oils or sterol additives.
Closing takeaway
What looks good on a cholesterol panel (lower LDL-C) may not reflect what’s happening inside your arteries. Think mechanistically: a truly cardioprotective fat should support membrane stability, resist oxidation, and help preserve clean, non-thrombogenic blood — properties more consistent with whole-food fats and balanced omega-3:6 intake than with highly oxidisable seed oils or added plant sterols.
📢 A Note on "Living Science"
Science is not a static destination; it is a moving target. While the principles discussed here are grounded in decades of metabolic research, new peer-reviewed data emerges every day, I am committed to accuracy.
I am committed to accuracy. If you are a researcher, clinician, or dedicated student of physiology and you find a piece of data here that does not align with the latest high-quality evidence, please reach out. I welcome civil, evidence-based corrections. My goal is to keep this resource as the most accurate "No-Nonsense" guide to Cardio Vascular Health on the internet. Let’s get better together.
*Disclaimer:
The information provided in this blog is for educational and informational purposes only and should not be construed as medical advice. While every effort is made to ensure accuracy, the content is not intended to replace professional medical consultation, diagnosis, or treatment. Always seek the guidance of a qualified healthcare provider with any questions regarding your health, medical conditions, or treatment options.
The author is not responsible for any health consequences that may result from following the information provided. Any lifestyle, dietary, or medical decisions should be made in consultation with a licensed medical professional.
If you have a medical emergency, please contact a healthcare provider or call emergency services immediately.


Comments